|
|
Post by Admin on Jan 13, 2021 4:00:09 GMT
A novel ACE2 isoform is expressed in human respiratory epithelia and is upregulated in response to interferons and RNA respiratory virus infection
Cornelia Blume, Claire L. Jackson, […]Vito Mennella
Nature Genetics (2021)
Abstract
Angiotensin-converting enzyme 2 (ACE2) is the main entry point in airway epithelial cells for SARS-CoV-2. ACE2 binding to the SARS-CoV-2 protein spike triggers viral fusion with the cell plasma membrane, resulting in viral RNA genome delivery into the host. Despite ACE2’s critical role in SARS-CoV-2 infection, full understanding of ACE2 expression, including in response to viral infection, remains unclear. ACE2 was thought to encode five transcripts and one protein of 805 amino acids. In the present study, we identify a novel short isoform of ACE2 expressed in the airway epithelium, the main site of SARS-CoV-2 infection. Short ACE2 is substantially upregulated in response to interferon stimulation and rhinovirus infection, but not SARS-CoV-2 infection. This short isoform lacks SARS-CoV-2 spike high-affinity binding sites and, altogether, our data are consistent with a model where short ACE2 is unlikely to directly contribute to host susceptibility to SARS-CoV-2 infection.
Main
With more than 53 million confirmed cases of COVID-19 and 1.3 million associated deaths worldwide (World Health Organization, 13 November 2020), there is an urgent need to understand the molecular mechanism of infection and disease to identify patients’ susceptibilities and targets for therapeutic intervention.
ACE2 is the main viral entry point for coronavirus N63, SARS-CoV and SARS-CoV-2, which cause severe acute respiratory syndromes, the last being responsible for COVID-19 in humans1,2,3,4. ACE2 binds to the S1 domain of trimeric SARS-CoV spike (S) glycoprotein1 and SARS-CoV-2 S protein5, which is primed by TMPRSS2 (ref. 6). Cellular entry of SARS-CoV is dependent on the extracellular domain of ACE2 being cleaved by TMPRSS2 protease at Arg 697 and Lys 716, and the transmembrane domain of ACE2 internalized with the virus7,8,9.
ACE2 is a carboxypeptidase with several known physiological functions including regulation of blood pressure, salt and water balance in mammals10,11, amino acid uptake in the small intestine12,13, and glucose homeostasis and pancreatic β-cell function14,15. Interestingly, ACE2 has been suggested to play an important role in protection from acute lung injury16,17,18,19.
ACE2 expression in different tissues is controlled by multiple promoter elements20,21,22. In human nasal epithelia and lung tissue, ACE2 expression has been reported to be interferon (IFN) regulated, with evidence of STAT1-, STAT3-, IRF8- and IRF1-binding sites within the ACE2 promoter23. Activation of IFN-responsive genes is an important antiviral defense pathway in humans, and both interferon and influenza exposure have been reported to increase ACE2 expression in the human airway23.
Bulk and single-cell RNA-sequencing (scRNA-seq) data24 detect low-level expression of ACE2 in multiple tissues25. ACE2 expression in the airways is relatively high in nasal epithelium and progressively lower in the bronchial and alveolar regions; this expression profile correlates with levels of infection of SARS-CoV-2 isolates from patients in different airway compartments26. Consistently, SARS-CoV-2 viral loads have been found to be higher in swabs taken from the nose than swabs taken from the throat of COVID-19 patients27. Highest ACE2 expression is seen in goblet and ciliated cells of the nasal epithelium25, and ACE2 protein localizes to the membrane of motile cilia of respiratory tract epithelia28. Consistent with this, SARS-CoV-2 has been detected in situ in ciliated airway cells and upper airway epithelium, in addition to pulmonary pneumocytes, in COVID-19 patients examined post mortem29. Airway multiciliated cells appear to be one of the main targets of SARS-CoV-2 infection26, and it has been demonstrated that SARS-CoV infection occurs through ACE2 on cilia in airway epithelia28. Altogether, these studies have established the upper airway as the main site of SARS-CoV-2 infection.
In the present study, we detail the identification of a new isoform of ACE2, which we name short ACE2, that is expressed in human nasal and bronchial respiratory epithelia, the main site of SARS-CoV-2 infection, and is preferentially expressed in asthmatic bronchial epithelium relative to full-length ACE2 (long ACE2). In primary airway cells, short ACE2 is upregulated in response to IFN treatment and infection with rhinovirus, but not SARS-CoV-2.
|
|
|
|
Post by Admin on Jan 13, 2021 20:54:07 GMT
Results Identification of new short ACE2 transcript We analyzed the expression of ACE2 in airway epithelia in our existing RNA-seq datasets obtained from nasal brushings and nasal epithelium cultured at the air–liquid interface (ALI). Visual analysis of alignment to the ACE2 gene region identified multiple reads mapping to a genomic region between exons 9 and 10 of the GENCODE v.33 ACE2 gene build with a discrete 3′-junction at GRCh38 chrX:15580281, suggesting a splice junction with downstream exon 10. Variable 5′-length indicated no splicing upstream of exon 8, suggesting a new ACE2 transcript starting with a new exon, which we call exon 9a, in the airway (Fig. 1a). Analysis of these RNA-seq data, with code developed by Cummings et al.30, also independently detected a new splice junction at chrX:15580281. Analysis of splice junctions identified by STAR31 aligner confirmed multiple uniquely mapped reads to a new exon/exon boundary removing a new intron of coordinates GRCh38 chrX:15578316–15580280. Assembly of transcriptomes using the SCALLOP tool independently identified this novel ACE2 transcript, including exon 9a to exon 19 (Fig. 1a). Analysis of relative expression of the short and full-length (long) ACE2 transcripts in these RNA-seq data showed that, in primary nasal epithelial cells, short ACE2 expression is significantly higher than long ACE2 expression (Fig. 1b). Study of the sequence of the exon 9a/intron boundary showed a strong U1-dependent consensus splice site sequence (AG|GTAAGTA), suggesting that it is a strong splice donor site (Fig. 1a). This splicing event introduces a new, in-frame ATG start codon 29 nucleotides (nt) upstream of the splice site (Fig. 1a), and a TATA box 148 nt upstream of the splice site (Fig. 1a), suggesting that this transcript is protein coding. Furthermore, a promoter flanking region has been identified at GRCh38 chrX:15581200–15579724 (ENSR00000902026), suggesting active transcription upstream of exon 9a (approximately chrX:15580402–chrX:15580281). Analysis of this region shows a near-consensus ISGF-3-binding site (TgGTTTCAgTTTCCt)32 159 bp upstream of the splice junction, a near-consensus AP-1-binding site (TGtGTCA)33 223 bp upstream of the splice site and an NF-κB-binding site (GGGTTTTCCC)34 787 bp upstream of the splice junction. This suggests that short ACE2 is under independent transcriptional control from full-length ACE2 expression, and that this may be controlled by IFN, AP-1 and NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) elements. Fig. 1: A novel short transcript of ACE2 is expressed in airway epithelia. 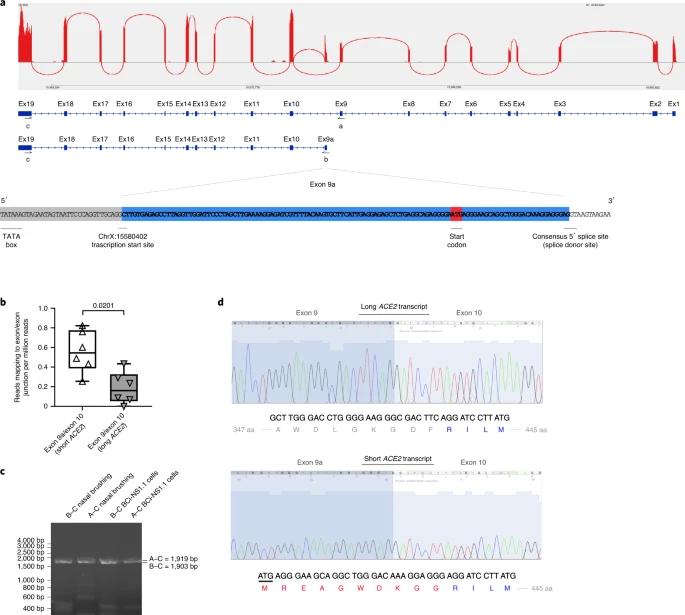 a, Sashimi plot showing splice junction between exons (Ex) 9 and 10, and between exons 9a and 10, counted from RNA-seq reads from one nasal brushing sample. GENCODE v.33 gene build exons and new SCALLOP transcriptome build exons, showing new exon 9a in a new transcript encompassing exons 9a–19, assembled by SCALLOP tool. Letters ‘A’, ‘B’ and ‘C’ indicate locations of primers for RT–PCR (Fig. 1c). Nucleotide sequence of novel exon 9a, plus 5′ UTR, start codon and splice junction, are shown. b, Box and whisker plot showing expression levels of short and long ACE2 transcripts in primary NECs (reads mapped to exon 9a/10 or exon 9/10 per million mapped reads) (P = 0.0201, paired, two-way Student’s t-test; n = 6 donors). c, Agarose gel electrophoresis image of long-range, transcript-specific PCR products amplifying full short ACE2 transcript and exons 9–19 of long ACE2 transcript from nasal epithelial brushings and BCi-NS1.1 cells, using different pairs of primers (A and C or B and C), specific to regions of ACE2 shown in a. d, Sanger sequencing electropherogram traces showing sequence at exon/exon boundaries of long ACE2 transcript exons 9–10 and short ACE2 transcript exons 9a–10. Amino acid (aa) translation is shown below. Expression of this new transcript was confirmed by reverse transcriptase (RT)–PCR using primers specific to exons 1, 9a and 19 and complementary DNA from both nasal brushings and differentiated immortalized bronchial epithelial cells, BCi-NS1.1, which differentiate robustly into airway multiciliated cells35 (Fig. 1c). Sanger sequencing confirmed the identity of these PCR amplicons (Fig. 1d). We then designed specific quantitative PCR (qPCR) primers to amplify the short and long transcripts of ACE2 individually, as well as primers to amplify both transcripts and to quantify total levels of ACE2 expression (Extended Data Fig. 1a,b). Expression of long ACE2 was confirmed in a number of cell lines and primary airway cells, with the highest expression being observed in the Vero E6 cell line, differentiated BCi-NS1.1 cells and in vitro differentiated nasal epithelial cells grown at ALI, with expression comparable to that observed in ex vivo nasal epithelial cells (NECs) (Fig. 2a,b). Expression of short ACE2 was low in Vero E6, HEK293, Caco2, RPE1, H441 and 16HBE cells, and this contrasted with differentiated BCi-NS1.1 cells and ex vivo or in vitro differentiated nasal cells, which exhibited high expression of this new isoform. Both isoforms were also expressed robustly in ex vivo or in vitro differentiated primary bronchial cells (Fig. 2a,b). Assessment of ACE2 isoform expression during differentiation of NECs grown at ALI in vitro showed very low ACE2 expression on day 1, with expression of both isoforms reaching levels comparable with those observed in primary nasal brushings at day 4 of ALI culture, when the start of cilia gene transcription is observed (usually days 4–7), being maintained until day 63 and reducing at day 84 as the cultures became senescent (Fig. 2c and Extended Data Fig. 2). This is consistent with published work showing that ACE2 expression (and SARS-CoV-2 infection) is dependent on airway epithelial cell differentiation36. Given reports that bronchial epithelial cells express lower levels of ACE2 than nasal cells23, we also compared expression of the long and short isoforms of ACE2 in these two cell types from multiple donors of primary tissue. Consistent with previous reports, total levels of ACE2 were lower in bronchial epithelial cells, which was due to reduced expression of both long and short forms of ACE2 (Fig. 2d). Fig. 2: Short ACE2 is expressed in different cell types. 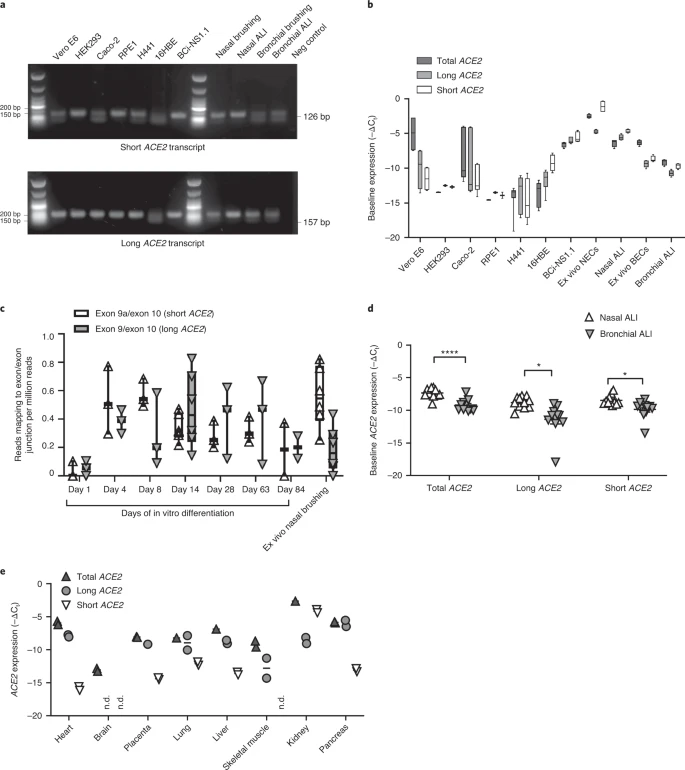 a, Representative agarose gel electrophoresis image of transcript-specific ACE2 RT–PCRs (n = 3) from different cell types. Size standard = NEB low-molecular-mass ladder. b, Box and whisker plots showing 5th to 95th percentiles (whiskers), median and quartiles (box) of baseline expression (−ΔCt) of total ACE2 (dark gray), long ACE2 (light gray) and short ACE2 (white) in cell lines and airway cells. Analysis was done at least in duplicate and from different passages or donors in all lines except RPE1 and HEK293 cells (analysis done in duplicate from one passage). c, Box and whisker plots showing 5th to 95th percentiles (whiskers), median and quartiles (box), plus individual data points showing relative expression (reads mapped to exon/exon boundary per million mapped reads) of short ACE2 transcript (white upward-pointing triangles) and long ACE2 transcript (gray downward-pointing triangles) in RNA-seq data from NECs at different stages of differentiation at ALI (days 1, 4, 8, 14, 28, 63 and 84; n = 3 donors for each time point) and primary ex vivo nasal brushings (n = 6 donors). d, Medians and individual data points showing baseline expression (−ΔCt) of total ACE2 transcripts, long ACE2 transcript and short ACE2 transcript in nasal (white upward-pointing triangles, n = 11 donors) and bronchial (gray downward-pointing triangles, n = 11 donors) ALI cultures from healthy donors, as determined using transcript-specific qPCR. Data were analyzed using the Mann–Whitney U-test. e, Medians and individual data points showing baseline expression (−ΔCt) of total ACE2 (dark gray upward-pointing triangles), long ACE2 (light gray circles) and short ACE2 (white downward-pointing triangles) transcript in a multiple tissue cDNA panel 1 (636742, TakaraBio), as determined using transcript-specific, probe-based ACE2 RT–qPCR. ND, not detected; n = 2 independent experiments. |
|
|
|
Post by Admin on Jan 14, 2021 0:32:34 GMT
Short ACE2 transcript is expressed in multiple tissues Transcript-specific, probe-based qPCR on cDNA from a multiple tissue control panel showed robust expression of the long transcript of ACE2 in all tissues tested except whole brain (Fig. 2e). The short transcript of ACE2 was detected in all tissues except whole brain and skeletal muscle, although the expression level was low in most tissues, with the highest expression in lungs and kidneys (Fig. 2e). Short ACE2 lacks most of the SARS-CoV-2-binding interface We modeled the structure of the predicted translation product of this short transcript of ACE2, based on the structure of full-length long ACE2 protein as a complex with the receptor-binding domain of SARS-CoV-2 resolved by cryo-electron microscopy (cryo-EM) (Protein Data Bank (PDB) 6M17)4. This analysis highlighted the extent of loss of the SARS-CoV-2-binding region in the predicted protein product of short ACE2, with many residues previously shown to be important for viral binding not present in this short ACE2 protein (Extended Data Fig. 3a–c). Assuming that the homologous parts of short ACE2 fold in the same way as full-length ACE2, molecular dynamic simulation of a short ACE2 homodimer suggested that it may form a stable structure (Extended Data Fig. 3d–f and Supplementary Video 1). Short ACE2 is detected in differentiated airway epithelia To investigate whether short ACE2 encodes a protein isoform in airway epithelial cells and other cell types, we performed western blotting analysis of cell lysates using multiple antibodies to ACE2 recognizing epitopes on different regions of the protein (Fig. 3a). Initially, we tested an antibody raised to the C-terminal domain (CTD) of ACE2 (anti-ACE2 CTD), which we anticipated would recognize a common epitope in long and short ACE2. Western blotting of lysates prepared from seven cell lines including Vero cells, which are used for infection assays of SARS-CoV37 and SARS-CoV-2 (ref. 38), identified 2 bands at 100 and 120 kDa, consistent with the presence of glycosylated and nonglycosylated forms of full-length (long) ACE2 protein1 (see below). We also detected an additional band at ~52 kDa, the expected molecular mass of short ACE2, in differentiated BCi-NS1.1 cells (Fig. 3b), and fully differentiated primary nasal and bronchial epithelial cultures (Fig. 3c). Expression of this protein in other cell lines was low, consistent with the qPCR data (Fig. 3b). Lack of correlation between long ACE2 protein levels and the intensity of the 52-kDa band suggests that the latter is not a degradation product of long ACE2. Preadsorption of the anti-ACE2 CTD antibody with the immunizing peptide, but not a peptide with a similar charge, blocked detection of both long and short ACE2 isoforms in the airway cells, confirming specific detection of short ACE2 (Fig. 3c). Fig. 3: Short ACE2 protein is expressed and not enriched on motile cilia relative to long ACE2 protein. 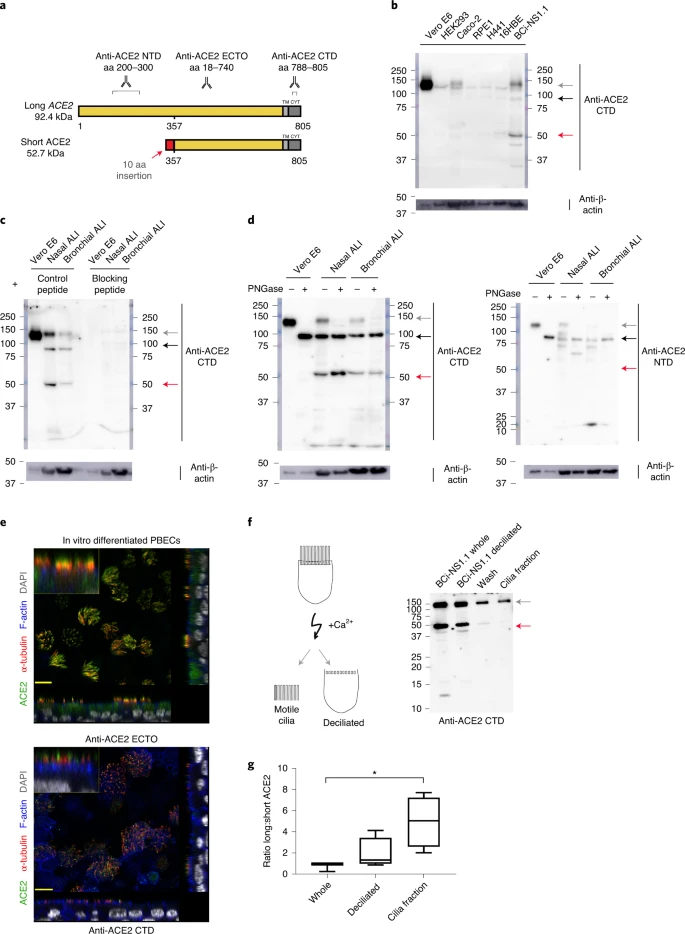 To orthogonally validate antibody specificity, and examine glycosylation of the ACE2 isoforms, we performed additional western blot analyses on cell lysates of both nasal and bronchial epithelial ALI cultures, as well as Vero E6 cells before and after treatment with PNGase F, an enzyme that removes N-linked oligosaccharides from glycoproteins. This confirmed that the ~120-kDa band detected by the anti-ACE2 CTD antibody was glycosylated ACE2 whereas the ~100-kDa band was non (or partially) glycosylated ACE2; the mobility of the ~52-kDa band corresponding to short ACE2 did not change, suggesting that it is not N-glycosylated. Similar results were obtained with an antibody raised against the ectodomain of ACE2 (anti-ACE2 ECTO; amino acids 18–740), which recognized glycosylated and nonglycosylated isoforms of long ACE2, as well as short ACE2. In contrast, an ACE2 N-terminal domain antibody (anti-ACE2 NTD) raised against amino acids 200–300, which are present only in the N-terminal region of long ACE2 recognized glycosylated and nonglycosylated isoforms of long ACE2 but, as expected, it did not recognize short ACE2 (Fig. 3d). To examine the localization of the ACE2 isoforms in airway epithelial cells, we performed immunofluorescence staining of differentiated ALI cultures of primary bronchial epithelial cells (PBECs) with anti-ACE2 antibodies visualized by confocal microscopy (Fig. 3e and Extended Data Fig. 4a–d). The antibodies recognizing common epitopes in short and long ACE2 showed localization mainly to the cellular apical regions and motile cilia. Thus, to further investigate whether short ACE2 localizes to cilia, as has been reported for full-length ACE2 (ref. 39), we purified motile cilia from BCi-NS1.1 cells. Consistent with our previous experiments, western blotting of whole cell lysates with anti-ACE2 CTD antibody showed a distinct band around 52 kDa, in addition to full-length ACE2 (Fig. 3f). Notably, the band corresponding to short ACE2 was not enriched in the cilia fraction albeit still present in detectable amounts (Fig. 3f), suggesting that it is predominantly localized to the apical regions of the cells. Densitometric analysis confirmed enrichment of long ACE2 in the cilia fraction relative to short ACE2 (Fig. 3g). Cell-free protein synthesis assays of the ectodomains of long and short ACE2 confirmed that both constructs were expressed at comparable levels with little evidence of degradation (Extended Data Fig. 5a). However, no expression of a green fluorescent protein (GFP)-tagged short ACE2 construct was seen in mammalian cell lines except in the H441 airway cell line, suggesting that the stability of short ACE2 may be cell type dependent (Extended Data Fig. 5b). INFs and RV infection upregulate short ACE2 To begin investigating the functional relevance of short ACE2 transcript expression, we first assessed whether it is an IFN-stimulated gene. As expected, treatment of bronchial epithelial cells with type I, II or III IFNs caused upregulation of MX1 and IP10 (Extended Data Fig. 6a,b) and upregulation of total ACE2 (Fig. 4a), which was largely due to an effect on short ACE2 rather than long ACE2 (Fig. 4a). In these experiments we observed effects of IFN-β > IFN-γ and IFN-λ > IFN-α in induction of short ACE2 expression (Fig. 4a); we also noted inhibitory effects of some of the IFNs on expression of long ACE2 (Extended Data Fig. 6c), such that only IFN-β caused a significant change in the ratio of short ACE2 relative to long ACE2 expression (Fig. 4a and Extended Data Fig. 6d). Based on the greater potency of IFN-β, we confirmed the induction of short ACE2 in differentiated bronchial epithelial cells treated with IFN-β (Extended Data Fig. 6e,f). Fig. 4: Short ACE2 is upregulated in response to IFN and RV16 infection but not SARS-CoV-2 infection. 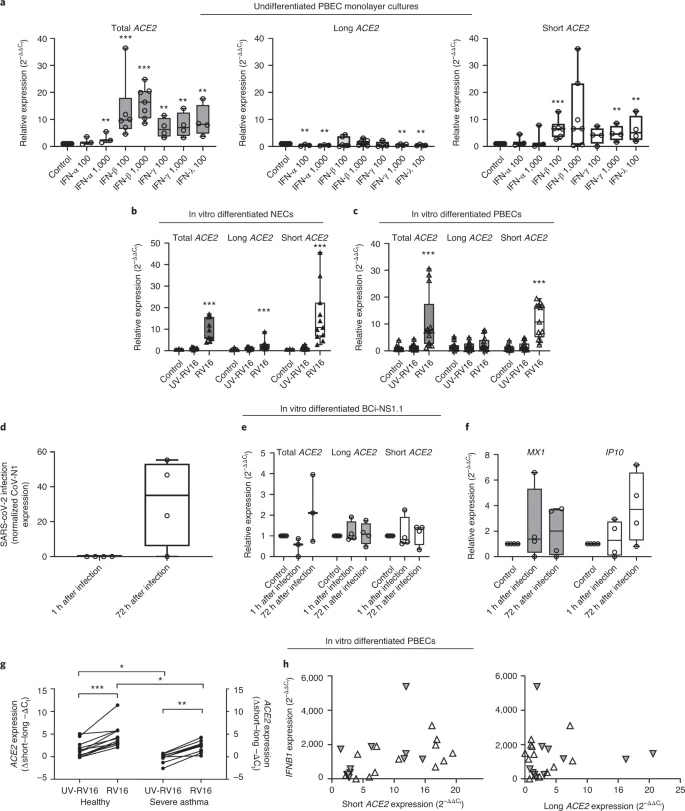 These data led us to evaluate the response to viral infection, because it has been reported that ACE2 expression is upregulated in this condition23. We exposed NECs grown at ALI to rhinovirus (RV16) and harvested cells 24 h after infection. The qPCR analysis showed a significant upregulation of both long and short ACE2 expression relative to UV-RV-treated controls (Fig. 4b). Although this is consistent with previously published work showing that ACE2 is upregulated in response to a ninefold increase in expression of short ACE2 compared with around a 2.5-fold increase in long ACE2 expression (Fig. 4b), parallel experiments using bronchial epithelial cells infected with RV16 confirmed that induction of short ACE2 but had no significant effect on long ACE2 (Fig. 4c). We next sought to investigate the effect of SARS-CoV-2 infection on the expression of the short transcript of ACE2. Differentiated BCi-NS1.1 cells were infected with SARS-CoV-2 and harvested at 1 and 72 h after infection (Fig. 4d); however, we did not observe a significant induction of either the long or the short isoforms of ACE2, or total ACE2 (Fig. 4e). Despite the fact that BCi-NS1.1 cells can respond to RV16 infection with IFN production and induction of IFN-stimulated genes MX1 and IP10 (Extended Data Fig. 6g,h), we did not observe significant induction of MX1 or CXCL10 in the SARS-CoV-2-infected cultures (Fig. 4f), consistent with previously published data demonstrating the ability of SARS-CoV-2 to inhibit both IFN expression and downstream signaling from the type I IFN receptor40,41,42,43. We next examined induction of short ACE2 in differentiated bronchial epithelial cultures from severe asthmatic donors who are known to have reduced type I and type III IFN responses to RV16 infection44,45. We observed a significant increase in expression of total ACE2 and short ACE2 but not long ACE2 when differentiated bronchial epithelial cultures from either healthy control individuals or people with severe asthma were infected with RV16 (Extended Data Fig. 7a,b). This resulted in a significant increase in the ratio of short ACE2 relative to long ACE2 (Fig. 4g and Extended Data Fig. 7c); however, for the severe asthma-derived cultures this increase was significantly less than that observed in cultures from healthy donors (Fig. 4g). The RV16-induced increase in short ACE2 expression was positively correlated with expression of IFNB1 (Fig. 4h), whereas there was no similar relationship for long ACE2 expression. Furthermore, as we observed previously44, RV-induced IFN-λ (interleukin (IL)-29/28) secretion was lower in the severe asthma-derived cultures (Extended Data Fig. 7d) and levels of IFN-λ were significantly correlated with expression levels of short ACE2 but not long ACE2 (Extended Data Fig. 7e). Finally, as ACE2 expression has been reported to be elevated in some patients with severe asthma who exhibit type I and II IFN signatures as a phenotypic trait46, we analyzed expression of long and short ACE2 in bronchial brushings from healthy controls and patients with severe asthma who were free of respiratory virus infections. This showed statistically significantly lower expression of total ACE2 and long ACE2 in patients with severe asthma, but no significant difference in short ACE2 expression between groups (Extended Data Fig. 7f). As a consequence, the ratio of short ACE2 to long ACE2 was significantly higher in patients with severe asthma (Extended Data Fig. 7g). In these severe cases, the expression of the IFN-stimulated genes OAS1 and ISG15 was significantly elevated (Extended Data Fig. 7h), and short ACE2 expression levels significantly correlated with IFN-γ protein levels in bronchoalveolar lavage (BAL) fluid (Extended Data Fig. 7i), suggesting that variation in expression of ACE2 isoforms in vivo may reflect the inflammatory status in asthma, independently of a virus-induced exacerbation. |
|
|
|
Post by Admin on Jan 14, 2021 20:34:57 GMT
Discussion
In the present study, we present identification and characterization of a short 11-exon transcript of human ACE2, consisting of a new previously unannotated (at the time of analysis) first exon, which we name exon 9a, and exons 10–19 of the long ACE2 transcript. We show that, although the long transcript of ACE2 is expressed in multiple tissues, this short ACE2 transcript is expressed predominantly in the airways. We show highest expression in primary respiratory epithelia, most notably in the nasal epithelium where the level of short transcript expression is higher than long ACE2 transcript expression. Expression of both transcripts of ACE2 depends on the differentiation of epithelial cells, with levels of expression comparable with primary nasal epithelia from days 4–63 of differentiation at ALI culture. We show that expression of this short transcript is regulated independently of the long transcript of ACE2, with putative promoter elements identified upstream of the transcriptional start site of the short ACE2 transcript. We confirm that this short transcript is translated into a protein product of around 52 kDa, which is not glycosylated in differentiated airway epithelial cells. Although the transcript lacks a signal peptide, recombinant protein expression studies confirm that the short ACE2 transcript can be translated into a protein product in vitro; however, within cells its expression appears to be relatively unstable. As is the case for other unstable proteins, this may be because the standard cell lines used for expression studies are not equipped to accomplish the required post-translational modifications or molecular folding of this molecule. In parallel with our studies, two independent reports identified the novel ACE2 transcript by extensive mining of public datasets47,48; however, endogenous protein expression of the short ACE2 isoform was not detected in a range of cell lines or undifferentiated PBECs by either study. Thus, detailed studies of short ACE2 function may require use of airway cell systems, especially differentiated cell models where short ACE2 appears more abundant, perhaps reflecting a stabilizing environment.
Previous studies have reported that ACE2 is an IFN-regulated gene and suggested that SARS-CoV-2 can exploit IFN-driven upregulation of ACE2 to enhance infection23. In our study, we show that it is the short transcript of ACE2 that is more strongly induced by type I, II and III IFNs and by viral infection than long ACE2, findings similar to two recent reports47,48. In our study, we performed detailed comparative dose–response studies and found that short ACE2 was not only upregulated by IFN-β and IFN-λ, reflecting innate epithelial responses to infection, but also by IFN-γ and, to a much lesser extent, IFN-α, suggesting a further contribution from co-resident immune cells such as T cells and plasmacytoid dendritic cells in vivo. Although we observed a small increase in long ACE2 expression in RV16-infected NECs, it seems unlikely that this was due to virus-induced IFN production, as we observed either no effect or an inhibitory effect of IFNs on long ACE2 expression. Given that viral infection also induces expression of cytokines such as IL-1β, which has been reported to induce ACE2 expression49, it seems more likely that cytokines other than IFNs induce long ACE2, the viral receptor form of ACE2. Given the absence of key residues required for SARS-CoV-2 spike binding and the evidence that short ACE2 cannot bind SARS-CoV-2 spike protein48, it is unlikely that IFNs have a detrimental effect in the airways by promoting SARS-CoV-2 entry, as has been suggested previously23. Such a conclusion is supported by a recent clinical trial of inhaled IFN-β in hospitalized patients who showed greater odds of clinical improvement and recovery, as well as reduced breathlessness compared with placebo50, thus highlighting the antiviral benefits of IFN-β.
Although patients with asthma have reduced susceptibility to SARS-CoV-2, and asthma symptoms are not exacerbated by SARS-CoV-2 infection51,52,53, it has been reported that ACE2 expression is higher in bronchial epithelium of a subset of asthma patients with type 2 low disease and characteristics resembling known risk factors for severe COVID-19 (ref. 46). As this increase in ACE2 expression was found to be associated with upregulation of viral response genes, it was suggested that therapies targeting the IFN family may be of benefit in this subset of patients46. Through analysis of ACE2 isoforms, our data show that long ACE2 expression is lower in people with severe asthma compared with healthy controls, even though we were able to identify upregulation of certain IFN-responsive genes in the asthma group. In contrast, expression of short ACE2 was comparable between the groups, and there was a higher ratio of expression of short ACE2:long ACE2 in severe asthma. It seems likely that this is linked to the inflammatory status of the airways and does not place people with asthma at greater risk of SARS-CoV-2 infection.
Of note, we did not observe induction of a short ACE2 in response to SARS-CoV-2 infection of differentiated airway epithelial cells, a model that closely mimics the route of infection in humans. This finding is similar to that reported for SARS-CoV-2 infection of Calu3 lung adenocarcinoma cells, and is distinct from responses in intestinal epithelial cells where short ACE2 was induced by SARS-CoV-2 infection48. These differences may reflect differences in infection levels and/or the ability of SARS-CoV2 to suppress IFN expression and signaling40,41,42,43; however the possibility of cell-type differences requires further consideration, especially as the airways are the primary route of SARS-CoV-2 infection.
The discovery of short ACE2 may have important consequences for the design of therapeutic approaches targeting ACE2 to tackle COVID-19 (refs. 54,55) and has implications for the numerous studies reporting on ACE2 expression levels and differences in levels of expression along airways, and across age groups and disease groups26,46,56,57 including COVID-19 disease severity57,58. This finding should be considered when selecting reagents for future studies of ACE2 expression in tissues relevant to SARS-CoV-2 viral infection, bearing in mind the genomic region targeted by primer sets and the epitopes recognized by antibodies.
In conclusion, we have identified short ACE2, a new isoform of ACE2 predominantly expressed in differentiated airway epithelial cells, especially in cells of the upper airways, which are the main site of SARS-CoV-2 infection. Our data suggest that the transcript encodes a 52-kDa protein that can be detected in airway epithelial cells, although, because it lacks a signal peptide, it may be a relatively unstable protein. We demonstrate that it is this isoform, rather than full-length ACE2, that is IFN regulated and inducible on RV infection. However, in conditions of IFN suppression, as observed during SARS-CoV-2 infection, or IFN-β deficiency, as in asthma, short ACE2 is not induced to the same degree as normal. Although the function of short ACE2 is unknown, its regulation by IFN suggests that it may play an essential role in innate antiviral defense mechanisms in the airways.
|
|

