Adaptive archaic hominin genes
As they migrated out of Africa and into Europe and Asia, anatomically modern humans interbred with archaic hominins, such as Neanderthals and Denisovans. The result of this genetic introgression on the recipient populations has been of considerable interest, especially in cases of selection for specific archaic genetic variants. Hsieh et al. characterized adaptive structural variants and copy number variants that are likely targets of positive selection in Melanesians. Focusing on population-specific regions of the genome that carry duplicated genes and show an excess of amino acid replacements provides evidence for one of the mechanisms by which genetic novelty can arise and result in differentiation between human genomes.

Copy number variants (CNVs) are subject to stronger selective pressure than single-nucleotide variants, but their roles in archaic introgression and adaptation have not been systematically investigated. We show that stratified CNVs are significantly associated with signatures of positive selection in Melanesians and provide evidence for adaptive introgression of large CNVs at chromosomes 16p11.2 and 8p21.3 from Denisovans and Neanderthals, respectively. Using long-read sequence data, we reconstruct the structure and complex evolutionary history of these polymorphisms and show that both encode positively selected genes absent from most human populations. Our results collectively suggest that large CNVs originating in archaic hominins and introgressed into modern humans have played an important role in local population adaptation and represent an insufficiently studied source of large-scale genetic variation.

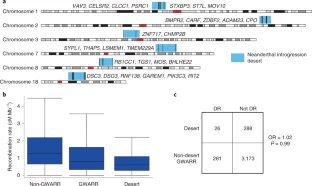
Fig. 1 Candidates for introgressed archaic and selective CNVs.
Joint distributions of P values for CNV stratification (x axis, Mann-Whitney U), archaic introgression (fD statistic, top two rows), and positive selection (population branch statistic, bottom row) tests. The archaic reference sequences used in the calculations of fD are Neanderthals (top row) and Denisovans (middle row), respectively. CNVs that show signatures of both positive selection and archaic introgression (red circles) are distinguished from loci that show signatures of introgression only (orange circles).
INTRODUCTION
Characterizing genetic variants underlying local adaptations in human populations is one of the central goals of evolutionary research. Most studies have focused on adaptive single-nucleotide variants that either arose as new beneficial mutations or were introduced after interbreeding with our now-extinct relatives, including Neanderthals and Denisovans. The adaptive role of copy number variants (CNVs), another well-known form of genomic variation generated through deletions or duplications that affect more base pairs in the genome, is less well understood, despite evidence that such mutations are subject to stronger selective pressures.

Fig. 2 Evidence for adaptive Denisovan introgression of the chromosome 16p12.2 duplication at 16p11.2 in Melanesians.
(A) Copy number (CN) estimates of DUP16p12 for the SGDP populations and three archaic samples. Pie charts indicate the CN frequency of populations (right) and the population fraction of CN genotypes (bottom). NDL, Neanderthals; DNS, Denisovans; SIB, Siberians; SA, South Asians; MEL, Melanesians; ME, Middle Easterners; EUR, Europeans; EA, East Asians; AMR, Native Americans; AFR, sub-Saharan Africans. (B) Geographic distribution for the DUP16p12 duplication genotypes of 242 independent blood-derived DNA samples from Melanesia. The CN color scheme matches that in (D). (C) FISH experiments using fosmid clones from 16p12.2 confirm an additional copy of DUP16p12 (red fosmid clone, 174222_ABC10_2_1_000044550500_M3 at 16p12.2; table S13; fig. S38) in a Melanesian cell line (GM10541, CN3) as opposed to a European cell line (GM12878, CN2). (D) (Top) Signals of adaptive introgression in the Melanesians at 16p11.2—the locus in which the DUP16p12 duplication was inserted. The heat map shows the CN distribution at chromosome 16p11.2. Fosmid clones (green: ABC10_000044688200_G16; blue: ABC10_000043626100_E12; table S13) indicate the region where the integration of DUP16p12 occurred at the 16p11.2 locus. (Bottom) PBS (left y axis) for SNVs (dots) and fD [horizontal lines, representing windows of 100 SNVs, computed using Denisovans as the archaic reference, right axis] at DUP16p12. Colored circles (blue) and/or horizontal lines (purple) indicate significant test statistics (P < 0.05). Note that introgression signals at both 16p12.2 and 16p11.2 disappear if Neanderthals are used as the archaic reference in the fD computation (figs. S36 and S47).
RATIONALE
This study focuses on the discovery of introgressed and adaptive CNVs that have become enriched in specific human populations. We combine whole-genome CNV calling and population genetic inference methods to discover CNVs and then assess signals of selection after controlling for demographic history. We examine 266 publicly available modern human genomes from the Simons Genome Diversity Project and genomes of three ancient hominins—a Denisovan, a Neanderthal from the Altai Mountains in Siberia, and a Neanderthal from Croatia. We apply long-read sequencing methods to sequence-resolve complex CNVs of interest specifically in the Melanesians—an Oceanian population distributed from Papua New Guinea to as far east as the islands of Fiji and known to harbor some of the greatest amounts of Neanderthal and Denisovan ancestry.

Fig. 3 Reconstruction of the structure and evolutionary history for the Melanesian–Denisovan duplication at chromosome 16p11.2.
(A) Structural comparison of chromosome 16p11.2 (human genome reference GRCh37; top), the structure-resolved Melanesian contig (middle) at 16p11.2, and the ancestral locus of DUP16p12 at 16p12.2 (KV880768.1; NCBI BioProject: PRJNA31257; bottom). Colored boxes denote annotated human segmental duplications, and lines connecting the sequences show regions of homology. Duplicated segments specific to the Melanesian genome (red dashed box) are indicated if derived from unique (colored arrows) or previous duplication (colored rectangles) sequences. The region of recurrent genome rearrangements associated with autism is highlighted (pink shaded area). (B) Schematic model for the evolution of the DUP16p12 duplication. The schematic depicts structural changes over time, leading to the Melanesian architecture. Evolutionary timing was estimated on the basis of a series of phylogenetic analyses using structure-resolved sequences from 16p12.2 and 16p11.2 loci (31). The absence of intermediate genomes makes the order of some structural changes uncertain. (C) A new member of the NPIPB gene family, NPIPB16 (1206 amino acids), in the Melanesian DUP16p12 sequence with predicted sites of positive selection. dN/dS analyses show positively selected amino acid substitutions at NPIPB16 lineages (blue circles) compared with other NPIPB genes. Note that the cluster of massive amino acid changes (red circles) at position 1236 to 1284 (alignment space) is predicted to result from two indel events in the C terminus of NPIPB16 as opposed to a series of independent amino acid substitution events (fig. S51).
RESULTS
Consistent with the hypothesis of archaic introgression outside Africa, we find a significant excess of CNV sharing between modern non-African populations and archaic hominins (P = 0.039). Among Melanesians, we observe an enrichment of CNVs with potential signals of positive selection (n = 37 CNVs), of which 19 CNVs likely introgressed from archaic hominins. We show that Melanesian-stratified CNVs are significantly associated with signals of positive selection (P = 0.0323). Many map near or within genes associated with metabolism (e.g., ACOT1 and ACOT2), development and cell cycle or signaling (e.g., TNFRSF10D and CDK11A and CDK11B), or immune response (e.g., IFNLR1). We characterize two of the largest and most complex CNVs on chromosomes 16p11.2 and 8p21.3 that introgressed from Denisovans and Neanderthals, respectively, and are absent from most other human populations. At chromosome 16p11.2, we sequence-resolve a large duplication of >383 thousand base pairs (kbp) that originated from Denisovans and introgressed into the ancestral Melanesian population 60,000 to 170,000 years ago. This large duplication occurs at high frequency (>79%) in diverse Melanesian groups, shows signatures of positive selection, and maps adjacent to Homo sapiens–specific duplications that predispose to rearrangements associated with autism. On chromosome 8p21.3, we identify a Melanesian haplotype that carries two CNVs, a ~6-kbp deletion, and a ~38-kbp duplication, with a Neanderthal origin and that introgressed into non-Africans 40,000 to 120,000 years ago. This CNV haplotype occurs at high frequency (44%) and shows signals consistent with a partial selective sweep in Melanesians. Using long-read sequencing genomic and transcriptomic data, we reconstruct the structure and complex evolutionary history for these two CNVs and discover previously undescribed duplicated genes (TNFRSF10D1, TNFRSF10D2, and NPIPB16) that show an excess of amino acid replacements consistent with the action of positive selection.

Fig. 4 Highly stratified CNVs at 8p21.3 in Melanesians and evidence for gene duplication and fusion followed by adaptive evolution at the TNFRSF10D locus.
(A) Manhattan plot for the P values of window-based FST test. The horizontal dashed line indicates the genome-wide Bonferroni-corrected significance. (B) (Top) Distribution of heterozygous sites (short black vertical bars) for a subset of the SGDP samples. The gray box at the top shows the location of DELMEL-NDL, whereas the blue-red box indicates the derived TNFRSF10D form, a fusion of TNFRSF10D1 (blue) and TNFRSF10D2 (red), as shown in Fig. 3C. (Bottom) Distributions of fD and PBS statistics, as well as CN trajectories of all samples across the region. (C) Comparison (Miropeats) of the major human allele versus chimpanzee genome structure, showing the tandem organization of the DUP10D variant and the predicted gene models. (D) Branch-site test of positive selection (dN/dS) using FLNC transcript data shows significant selection signals (P = 0.005) compared with the null model and a cluster of positively selected amino acid substitutions at the transmembrane domain of TNFRSF10D. Coding-sequence phylogeny shows significant positive selection (orange; dN/dS ratios > 1; P < 0.05) on specific branches. Note that the orangutan paralogous sequences form a single clade as a result of interlocus gene conversion (fig. S60).
CONCLUSION
Our results suggest that large CNVs originating in archaic hominins and introgressed into modern humans have played an important role in local population adaptation and represent an insufficiently studied source of large-scale genetic variation that is absent from current reference genomes.
Science 18 Oct 2019:
Vol. 366, Issue 6463, eaax2083