Post by Admin on Dec 15, 2020 22:17:48 GMT
Transcriptome-wide association study
We performed transcriptome-wide association study(TWAS)21,22
to link GWAS results to tissue-specific gene expression data by
inferring gene expression from known genetic variants that are
associated with transcript abundance (eQTL). For this analysis we
used GTEx v8 data for two disease-relevant tissues chosen a priori:
whole blood and lung (Figure 2). We selected genes with p<0.05
in these tissues and performed a combined meta-TWAS analysis,23
incorporating eQTL data from other tissues in GTEX, to optimise
power to detect differences in predicted expression in lung
or blood.
We discovered 5 genes with genome-wide significant differences in
predicted expression compared to controls (Supplementary Table 7).
This included 4 genes with differential predicted expression in lung
tissue (3 on chr3: CCR2, CCR3 and CXCR6, and one on chr5: MTA2B;
Supplementary Tables 8-10).
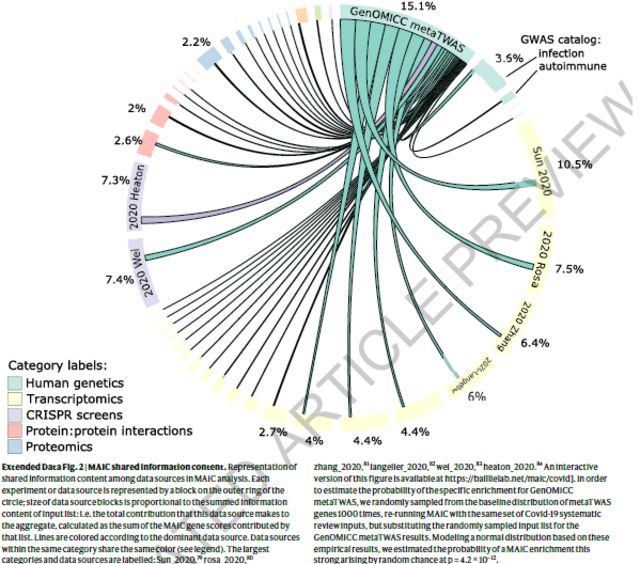
We used meta-analysis by information content (MAIC)24 to put
these results in the context of existing biological knowledge about
host-virus interactions in Covid. We combined the top 2000 genes
in metaTWAS with previous systematically-compiled experimental
evidence implicating human genes in SARS-CoV-2 replication and host
response. MAIC derives a data-driven weighting for each of a range
of experimental data sources in the form of gene lists, and outperforms
other approaches to providing a composite of multiple lists.24
We found that the GenOMICC TWAS results had greater overlap with
results from transcriptomic, proteomic and CRISPR studies of host
genes implicated in Covid-19 than any other data source (Extended
Data Figure 2).
Genetic correlations
We used the high-definition likelihood (HDL) method25 to provide an
initial estimate the SNP-based heritability (the proportion of phenotypic
variance that is captured by additive effects at common SNPs)
for severe Covid-19 to be 0.065 (SE = 0.019). We were not able to detect
a significant signal for heritability in two additional analyses: firstly,
using controls from the 100,000 genomes project (in which matching
to the GenOMICC cases is less close, which may limit heritability estimation)
and secondly, in a smaller GWAS comparing some GenOMICC
cases with UK Biobank controls, using matching of BMI and age where
possible. This second analysis was less powerful because of the lack of
close matches for many cases (ncases = 1260; ncontrols = 6300; Supplementary
Figure 14). Including rare variants in future analyses, with larger
numbers of cases, will provide a more comprehensive estimate of heritability.
We also tested for genetic correlations with other traits, that
is, the degree to which the underlying genetic components are shared
with severe Covid-19. Using the HDL method, we identified significant
negative genetic correlations with educational attainment and intelligence.
Significant positive genetic correlations were detected for a
number of adiposity phenotypes including body mass index and leg
fat (Supplementary Figure 19).
Consistent with GWAS results from other infectious and inflammatory
diseases, there was a significant enrichment of strongly associated
variants in promoters and enhancers,26 particularly those identified by
the EXaC study as under strong evolutionary selection (Supplementary
Figure 18).27 The strongest tissue type enrichment was in spleen
(which may reflect enrichment in immune cells), followed by pancreas
(Supplementary Figure 20).
We performed transcriptome-wide association study(TWAS)21,22
to link GWAS results to tissue-specific gene expression data by
inferring gene expression from known genetic variants that are
associated with transcript abundance (eQTL). For this analysis we
used GTEx v8 data for two disease-relevant tissues chosen a priori:
whole blood and lung (Figure 2). We selected genes with p<0.05
in these tissues and performed a combined meta-TWAS analysis,23
incorporating eQTL data from other tissues in GTEX, to optimise
power to detect differences in predicted expression in lung
or blood.
We discovered 5 genes with genome-wide significant differences in
predicted expression compared to controls (Supplementary Table 7).
This included 4 genes with differential predicted expression in lung
tissue (3 on chr3: CCR2, CCR3 and CXCR6, and one on chr5: MTA2B;
Supplementary Tables 8-10).
We used meta-analysis by information content (MAIC)24 to put
these results in the context of existing biological knowledge about
host-virus interactions in Covid. We combined the top 2000 genes
in metaTWAS with previous systematically-compiled experimental
evidence implicating human genes in SARS-CoV-2 replication and host
response. MAIC derives a data-driven weighting for each of a range
of experimental data sources in the form of gene lists, and outperforms
other approaches to providing a composite of multiple lists.24
We found that the GenOMICC TWAS results had greater overlap with
results from transcriptomic, proteomic and CRISPR studies of host
genes implicated in Covid-19 than any other data source (Extended
Data Figure 2).
Genetic correlations
We used the high-definition likelihood (HDL) method25 to provide an
initial estimate the SNP-based heritability (the proportion of phenotypic
variance that is captured by additive effects at common SNPs)
for severe Covid-19 to be 0.065 (SE = 0.019). We were not able to detect
a significant signal for heritability in two additional analyses: firstly,
using controls from the 100,000 genomes project (in which matching
to the GenOMICC cases is less close, which may limit heritability estimation)
and secondly, in a smaller GWAS comparing some GenOMICC
cases with UK Biobank controls, using matching of BMI and age where
possible. This second analysis was less powerful because of the lack of
close matches for many cases (ncases = 1260; ncontrols = 6300; Supplementary
Figure 14). Including rare variants in future analyses, with larger
numbers of cases, will provide a more comprehensive estimate of heritability.
We also tested for genetic correlations with other traits, that
is, the degree to which the underlying genetic components are shared
with severe Covid-19. Using the HDL method, we identified significant
negative genetic correlations with educational attainment and intelligence.
Significant positive genetic correlations were detected for a
number of adiposity phenotypes including body mass index and leg
fat (Supplementary Figure 19).
Consistent with GWAS results from other infectious and inflammatory
diseases, there was a significant enrichment of strongly associated
variants in promoters and enhancers,26 particularly those identified by
the EXaC study as under strong evolutionary selection (Supplementary
Figure 18).27 The strongest tissue type enrichment was in spleen
(which may reflect enrichment in immune cells), followed by pancreas
(Supplementary Figure 20).