|
|
Post by Admin on Apr 30, 2021 20:38:52 GMT
Figure 3: Distribution of ROH. 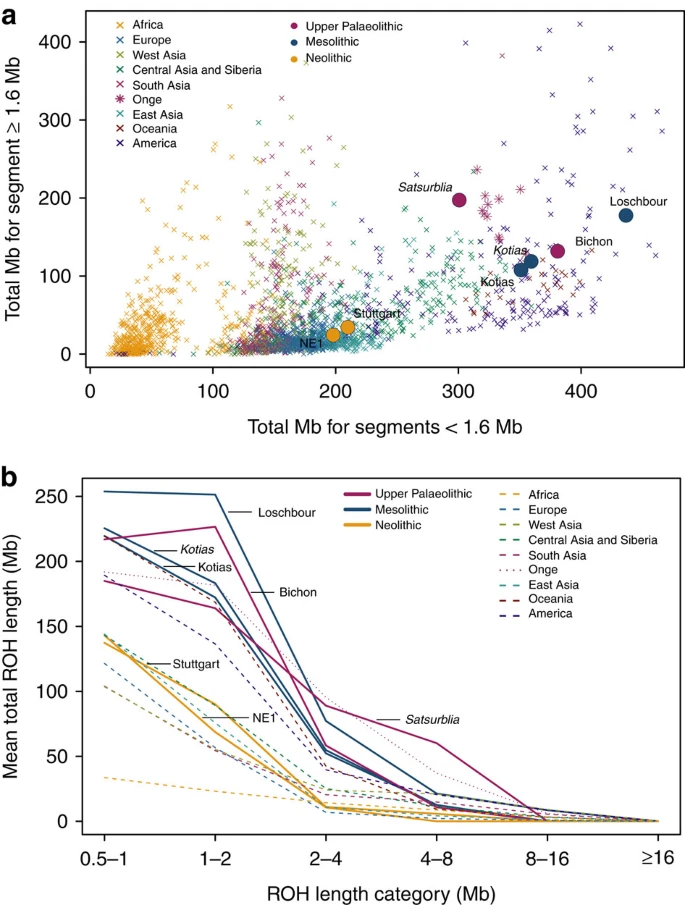 (a). The total length of short ROH (<1.6 Mb) plotted against the total length of long ROH (≥1.6 Mb) and (b) mean total ROH length for a range of length categories. ROH were calculated using a panel of 199,868 autosomal SNPs. For Kotias we analysed both high-coverage genotypes and genotypes imputed from downsampled data (marked in italics; see Supplementary Information). Diploid genotypes imputed from low-coverage variant calls were used for Satsurblia and high-coverage genotypes were used for all other samples. A clear distinction is visible between either WHG and CHG who display an excess of shorter ROH, akin to modern Oceanic and Onge populations, and EF who resemble other populations with sustained larger ancestral population sizes. Caucasus hunter-gatherer contribution to subsequent populations We next explored the extent to which Bichon and CHG contributed to contemporary populations using outgroup f3(African; modern, ancient) statistics, which measure the shared genetic history between an ancient genome and a modern population since they diverged from an African outgroup. Bichon, like younger WHG, shows strongest affinity to northern Europeans (Supplementary Fig. 3), while contemporary southern Caucasus populations are the closest to CHG (Fig. 4a and Supplementary Fig. 3), thus implying a degree of continuity in both regions stretching back at least 13,000 years to the late Upper Palaeolithic. Continuity in the Caucasus is also supported by the mitochondrial and Y chromosomal haplogroups of Kotias (H13c and J2a, respectively) and Satsurblia (K3 and J), which are all found at high frequencies in Georgia today22,23,24 (Supplementary Note 8). Figure 4: The relationship of Caucasus hunter-gatherers to modern populations. 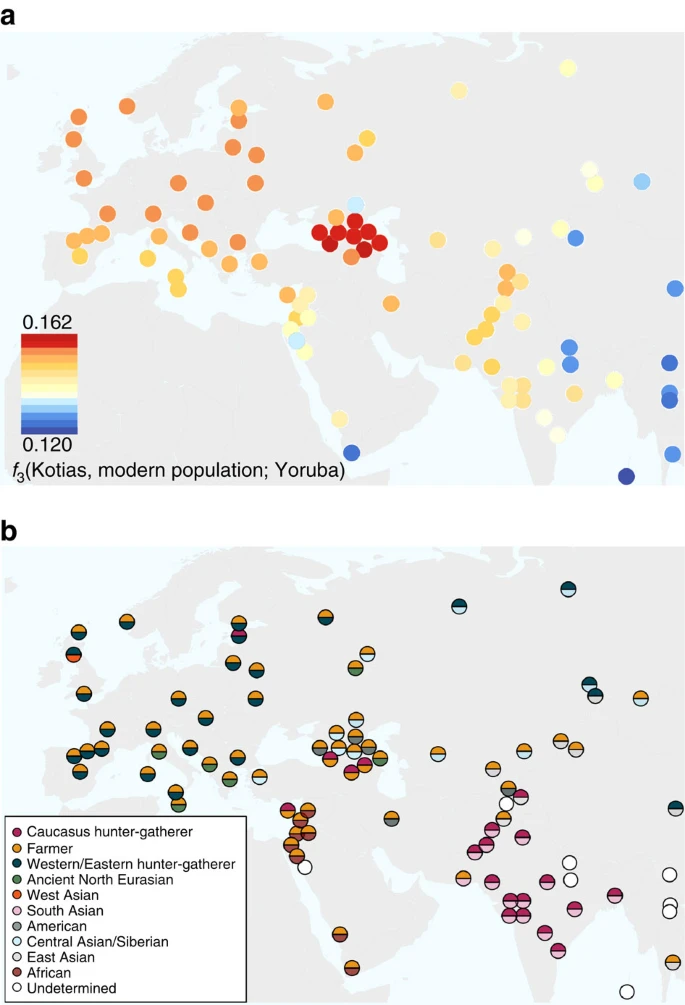 (a). Genomic affinity of modern populations1 to Kotias, quantified by the outgroup f3-statistics of the form f3(Kotias, modern population; Yoruba). Kotias shares the most genetic drift with populations from the Caucasus with high values also found for northern Europe and central Asia. (b). Sources of admixture into modern populations: semicircles indicate those that provide the most negative outgroup f3 statistic for that population. Populations for which a significantly negative statistic could not be determined are marked in white. Populations for which the ancient Caucasus genomes are best ancestral approximations include those of the Southern Caucasus and interestingly, South and Central Asia. Western Europe tends to be a mix of early farmers and western/eastern hunter-gatherers while Middle Eastern genomes are described as a mix of early farmers and Africans. EF share greater genetic affinity to populations from southern Europe than to those from northern Europe with an inverted pattern for WHG1,2,3,4,5. Surprisingly, we find that CHG influence is stronger in northern than Southern Europe (Fig. 4a and Supplementary Fig. 3A) despite the closer relationship between CHG and EF compared with WHG, suggesting an increase of CHG ancestry in Western Europeans subsequent to the early Neolithic period. We investigated this further using D-statistics of the form D(Yoruba, Kotias; EF, modern Western European population), which confirmed a significant introgression from CHG into modern northern European genomes after the early Neolithic period (Supplementary Fig. 4). |
|
|
|
Post by Admin on May 1, 2021 4:44:42 GMT
CHG origins of migrating Early Bronze Age herders We investigated the temporal stratigraphy of CHG influence by comparing these data to previously published ancient genomes. We find that CHG, or a population close to them, contributed to the genetic makeup of individuals from the Yamnaya culture, which have been implicated as vectors for the profound influx of Pontic steppe ancestry that spread westwards into Europe and east into central Asia with metallurgy, horseriding and probably Indo-European languages in the third millenium BC5,7. CHG ancestry in these groups is supported by ADMIXTURE analysis (Fig. 1b) and admixture f3-statistics14,25 (Fig. 5), which best describe the Yamnaya as a mix of CHG and Eastern European hunter-gatherers. The Yamnaya were semi-nomadic pastoralists, mainly dependent on stock-keeping but with some evidence for agriculture, including incorporation of a plow into one burial26. As such it is interesting that they lack an ancestral coefficient of the EF genome (Fig. 1b), which permeates through western European Neolithic and subsequent agricultural populations. During the Early Bronze Age, the Caucasus was in communication with the steppe, particularly via the Maikop culture27, which emerged in the first-half of the fourth millennium BC. The Maikop culture predated and, possibly with earlier southern influences, contributed to the formation of the adjacent Yamnaya culture that emerged further to the north and may be a candidate for the transmission of CHG ancestry. In the ADMIXTURE analysis of later ancient genomes (Fig. 1b) the Caucasus component gives a marker for the extension of Yamnaya admixture, with substantial contribution to both western and eastern Bronze Age samples. However, this is not completely coincident with metallurgy; Copper Age genomes from Northern Italy and Hungary show no contribution; neither does the earlier of two Hungarian Bronze Age individuals. Figure 5: Lowest admixture f3-statistics of the form f3 (X, Y; Yamnaya). 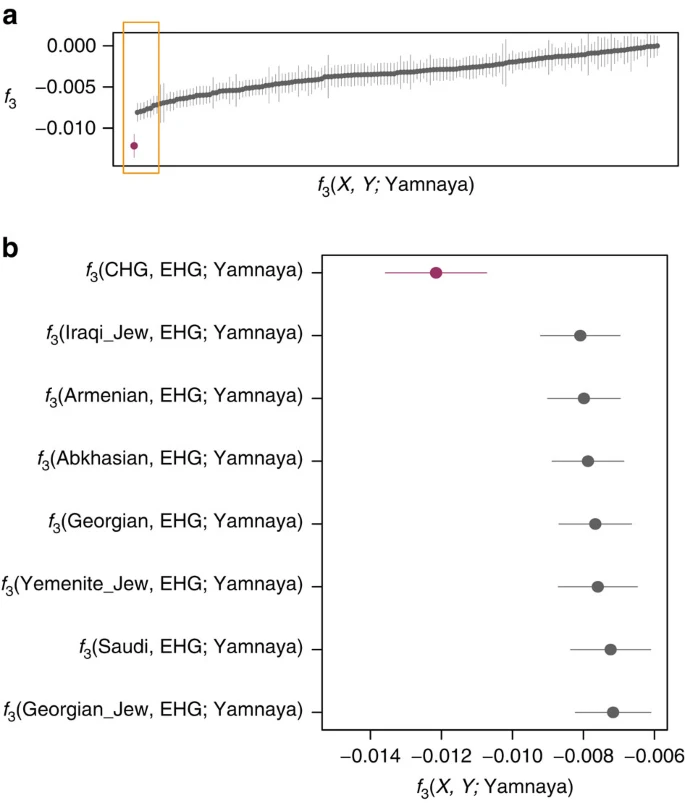 These statistics represent the Yamnaya as a mix of two populations with a more negative result signifying the more likely admixture event. (a). All negative statistics found for the test f3(X, Y; Yamnaya) with the most negative result f3(CHG, EHG; Yamnaya) highlighted in purple. Lines bisecting the points show the standard error. (b). The most significantly negative statistics which are highlighted by the yellow box in a. Greatest support is found for Yamnaya being a mix of Caucasus hunter-gatherers (CHG) and Russian hunter-gatherers who belong to an eastern extension of the WHG clade (EHG). Modern impact of CHG ancestry In modern populations, the impact of CHG also stretches beyond Europe to the east. Central and South Asian populations received genetic influx from CHG (or a population close to them), as shown by a prominent CHG component in ADMIXTURE (Supplementary Fig. 5; Supplementary Note 9) and admixture f3-statistics, which show many samples as a mix of CHG and another South Asian population (Fig. 4b; Supplementary Table 9). It has been proposed that modern Indians are a mixture of two ancestral components, an Ancestral North Indian component related to modern West Eurasians and an Ancestral South Indian component related more distantly to the Onge25; here Kotias proves the majority best surrogate for the former28,29 (Supplementary Table 10). It is estimated that this admixture in the ancestors of Indian populations occurred relatively recently, 1,900–4,200 years BP, and is possibly linked with migrations introducing Indo-European languages and Vedic religion to the region28. |
|
|
|
Post by Admin on May 1, 2021 21:05:53 GMT
Discussion
Given their geographic origin, it seems likely that CHG and EF are the descendants of early colonists from Africa who stopped south of the Caucasus, in an area stretching south to the Levant and possibly east towards Central and South Asia. WHG, on the other hand, are likely the descendants of a wave that expanded further into Europe. The separation of these populations is one that stretches back before the Holocene, as indicated by local continuity through the Late Palaeolithic/Mesolithic boundary and deep coalescence estimates, which date to around the LGM and earlier. Several analyses show that CHG are distinct from another inferred minor ancestral population, ANE, making them a divergent fourth strand of European ancestry that expands the model of the human colonization of that continent.
The separation between CHG and both EF and WHG ended during the Early Bronze Age when a major ancestral component linked to CHG was carried west by migrating herders from the Eurasian Steppe. The foundation group for this seismic change was the Yamnaya, who we estimate to owe half of their ancestry to CHG-linked sources. These sources may be linked to the Maikop culture, which predated the Yamnaya and was located further south, closer to the Southern Caucasus. Through the Yamanya, the CHG ancestral strand contributed to most modern European populations, especially in the northern part of the continent.
Finally, we found that CHG ancestry was also carried east to become a major contributor to the Ancestral North Indian component found in the Indian subcontinent. Exactly when the eastwards movement occurred is unknown, but it likely included migration around the same time as their contribution to the western European gene pool and may be linked with the spread of Indo-European languages. However, earlier movements associated with other developments such as that of cereal farming and herding are also plausible.
The discovery of CHG as a fourth ancestral component of the European gene pool underscores the importance of a dense geographical sampling of human palaeogenomes, especially among diverse geographical regions. Its separation from other European ancestral strands ended dramatically with the extensive population, linguistic and technological upheavals of the Early Bronze Age resulting in a wide impact of this ancestral strand on contemporary populations, stretching from the Atlantic to Central and South Asia.
|
|
|
|
Post by Admin on May 1, 2021 21:45:28 GMT
Supplementary Note 3
Kotias and Satsurblia form a clade with respect to other ancient samples while
Bichon shares close affinity to other western hunter-gatherers
D-statistics of the form D(Yoruba, OA; Satsurblia, Kotias) and D(Yoruba, OA; Bichon, WHG)
were used to assess whether the pairs of samples (Kotias, Satsurblia) and (Bichon, WHG) are
compatible with forming a clade in an unrooted tree with respect to an African outgroup and
other ancient samples (OA). For the test D(Yoruba, OA; Satsurblia, Kotias) we found most
statistics to have non-significant (zero) values (Supplementary Table 2) which supports Kotias
and Satsurblia forming a clade to the exclusion of other branches of ancient ancestry. The only
population for which a positive value was observed was the Sintasha Bronze Age culture. These
Uralic people are genetically similar to Corded Ware populations10 and this result could be
explained by the temporally closer Kotias representing a better donor for CHG ancestry than the
older Satsurblia for this population. These D-statistics confirm inferences from PCA and
ADMIXTURE that Kotias and Satsurblia are genetically distinct from other broadly
contemporaneous ancient genomes. When we performed the tests D(Yoruba, Satsurblia; OA,
Kotias) and D(Yoruba, Kotias; OA, Satsurblia) we found positive values of 0.07-0.16 with
corresponding Z-scores of 9.66-24.18. This shows that there is enough power to detect signals
of admixture using this dataset and that the zero values found above are not due to paucity of
data.
The test D(Yoruba, OA; Bichon, WHG) (where WHG were represented by the highest coverage
WHG genomes, Loschbour1 and La Braña6) resulted in insignificant values for 95% of tests
consistent with Bichon having more recent shared ancestry with WHG than with most other
ancient lineages (Supplementary Table 3). We consistently found zero-values when the OA
involved was an eastern hunter-gatherer (EHG), a CHG or a Pleistocene hunter-gatherer
showing that Bichon forms a clade with WHG to the exclusion of these other hunter-gatherer
groups. We did not always find zero-values however when we let OA be a Scandinavian huntergatherer (SHG; Supplementary Table 3). WHG are proposed to be part of a hunter-gatherer metapopulation, which also encompasses SHG and EHG, that ranged over northern Europe from as far west as Spain to as far east as Russia7. These three hunter-gatherer groups cannot be related by a simple tree as there are signals of admixture between these groups7.
This explains why Bichon does not always form a clade with other WHG to the exclusion of SHG.
When we inverted the statistics and evaluated D(Yoruba, Bichon; OA, WHG) and D(Yoruba,
WHG; OA, Bichon) we consistently found statistically significant values (Z >3). This shows that
admixture can be detected for the genotype coverage found in this dataset. We also found
similar results when we let the Hungarian sample KO12 represent WHG. It is interesting to note that Bichon, as well as other WHG, form a clade with both MA1 and EHG to the exclusion of CHG (Supplementary Table 6). This suggests the Ancient North Eurasian (ANE) ancestry and WHG ancestry may have shallower roots and diverged subsequent to splitting from CHG
(Supplementary Fig. 2). This is consistent with ADMIXTURE analysis and the geographic range
of these groups - CHG were separated from these North Eurasian hunter-gatherers by the
Caucasus mountain range.
We also explored the relationships between ancient samples by performing outgroup f3-statistics
of the form f3(X, OA; Yoruba) where we let X be Kotias, Satsurblia and Bichon in turn and OA be
all other ancient groups in the dataset (Supplementary Fig. 1). These statistics are informative
as their magnitude is proportional to the amount of shared genetic history between the ancient
individuals (X and OA) since they diverged from an African (in this case Yoruban) outgroup.
We found that CHG share the most drift with each other and the least drift with the Pleistocene
sample Ust’-Ishim (Supplementary Fig. 1A&B). Other ancient samples share an intermediate
amount of drift with no obvious pattern to the distribution of allele sharing. Bichon shares the
most genetic drift with other western hunter-gatherers, followed by Scandinavian and eastern
hunter-gatherers (Supplementary Fig. 1C). The fact that Bichon is closest to other WHG, and
not equally close to SHG and EHG, suggests that there may have already been sub-structure
between these hunter-gatherer groups 13,700 years ago when Bichon was alive.
|
|
|
|
Post by Admin on May 1, 2021 22:47:54 GMT
Supplementary Note 4
Caucasus hunter-gatherers and early farmers are sister groups with an earlier
divergence for western hunter-gatherers
To explore the topology between CHG, WHG and early farmers (EF) we used available high
coverage data and performed f3-statistics (see methods), attempting all possible triplet
combinations for these three groups (Figure 2A; Supplementary Table 4). When we did this we
presumed that two samples form a clade and the other sample is the outgroup to this clade. For
the correct topology we would expect f3 > 0, as the two correctly grouped samples will have
shared drift since they diverged from the outgroup. For incorrect topologies we would expect f3 =
0 as the incorrectly grouped samples will not have shared drift exclusive to themselves. We
found that f3(WHG, CHG; EF) tended to equal zero and gave the smallest values of all our
tests. This makes it unlikely that WHG and CHG are sister groups to the exclusion of EF. The
largest values were found for f3(CHG, EF; WHG) (Z > 14.4) suggesting that CHG and EF form a
clade to the exclusion of WHG. We did however also find positive statistics for the test f3(WHG,
EF; CHG) (Z > 8.5) but these were not as significant as for the former topology. WHG
introgression into EF has been proposed previously 1,2,4,5 and positive statistics for f3(WHG, EF;
CHG) could be a function of this admixture (admixture is also suggested by D-statistics of the
form D(Yoruba, WHG; CHG, EF) (Supplementary Table 8) and ADMIXTURE analysis (Figure
1B)). As the signal for EF and CHG forming a clade is much stronger than for the other two
topologies we consider the most parsimonious scenario to be that farmers and CHG are sister
groups that diverged from each other after splitting from WHG.
Unfortunately we did not have a high coverage diploid sample representing ANE to include in
these analyses. Analyses using D-statistics (Supplementary Table 6) revealed however that
ANE and WHG group together to the exclusion of CHG. It therefore seems likely that an ancient
south (Neolithic farmers and CHG) divergence from the ancient North (WHG and ANE) was the
earliest split for these groups. This is shown in Supplementary Fig. 2 which extends the model
proposed in 1 to include CHG. To fit this proposed model CHG and EF should form a clade to
the exclusion of Eastern non-Africans which is indeed supported by zero values for D(Yoruba,
eastern non-African, CHG, EF) (Supplementary Table 8). CHG and EF also form a clade to the
exclusion of ANE as represented by MA1 (Supplementary Table 8).
Supplementary Note 5
Dating the split among Caucasus hunter-gatherers, western hunter-gatherers and early
farmers
We used G-PhoCS54 to reconstruct the joint demographic history of western and Caucasus
hunter -gatherers (WHG and CHG respectively) and early farmers (EF). This analysis requires
(1) the topology of the underlying population tree; (2) sequence data from short, homologous
windows; and (3) specified directional gene flow between branches (migration bands).
Topology of population tree
G-PhoCS represents the demographic history of a collection of samples by a (binary) tree in
which each branch is a population, with each sample belonging to a different leaf branch and
interior branches corresponding to ancestral populations. To find the most likely topology of this
tree, we used f3 analysis to determine the most likely ordering of the population splits (see
Figure 2B for a graphical representation). For the G-PhoCS analyses, we considered both a tree
with only the ancient genomes, and a tree with an African San Pygmy55 as the outgroup.
Genome-wide windows of high sample coverage for demographic analyses Since this analysis requires sequence data from all genomes in short (1 kilobase (kb)) homologous windows, we chose high-coverage genomes to represent each group (Bichon and
Loschbour to represent WHG, Kotias to represent CHG, and either Stuttgart or NE1 to represent
EF). In addition, we used a high-quality San Pygmy genome55 as an outgroup. To find the best
set of windows, we first generated all-sites coverage information for chromosomes 1 to 22,
restricted to regions classified as “neutral” according to the filters in54 (using UCSC liftOver tool
to translate coordinates from hg18 to hg19), and extracted the depth information using a
program written in C, again filtering for sites with read depth between 10 and 35 (we avoid sites
with very low and very high coverage because alignment and genotyping is problematic (for
more details see 37):
samtools mpileup -C50 -uDI -f <reference.fa> -r <chromosome> \
-l <bedfile with accepted regions> <bamfiles> | bcftools view -gc – \
| get_depth_intervals minCover=10 maxCover=35
interval_file=<chromosome>
We then scanned each chromosome for good windows, using a simple heuristic to maximise the
sample coverage. We start by finding the first 1 kb window with at least 80% coverage. We then
search locally for a window within the next 10 kb for the 1 kb window with the highest coverage.
Finally, we jump 5 kb forward from the chosen location and repeat the process until we reach
the end of the chromosome. For the whole genome, this search yielded a total of 152,883 highquality windows. We then used SAMtools/BCFtools56 (using flags as above) and custom programs written in C and MATLAB to extract genotypes for the windows and converted the genotypes into fasta files for G-PhoCS. To deal with DNA damage in ancient samples, we “in
vitro” deaminated all our sequences, as already done for previous analyses of aDNA57.
Directional gene flow between branches
Because our WHG samples predate the arrival of farming to central and northern Europe58, any
gene flow creating shared drift between EF and WHG must be from WHG to EF. Ideally, we
would like our model to only allow gene flow between WHG and EF after the arrival of farming to
the WHG locations. However, G-PhoCS requires migration to start or stop at time where
populations split. Fortunately, our analysis puts the split between the two WHG, Bichon and
Loschbour, at around 14k years ago, just a few thousand years prior to farming. We therefore
allow gene flow between WHG and EF only after this split. Because Loschbour is temporally and
geographically closer than Bichon to the EF, we allow only gene flow from Loschbour to the EF.
Converting dates from ancient genomes
G-PhoCS assumes samples to be contemporaneous. The ages of our ancient genomes all fell
within a range of ~6k years (~7 kya for the youngest, EF, to ~13 kya for the oldest, Bichon). This
discrepancy is relatively small compared to the ages of the splits of interest, and will not affect
estimates in a qualitative way (especially given the size of the confidence interval of this type of
analysis). To convert split times for a given node as computed by G-PhoCS into calendar dates,
we added the mean of the ages of the samples that defined that node. The only modern
genome is the San, which is only used as an outgroup; as such, the age of that split between
the San and the ancient genomes is not of interest (and given how old that split is, a difference
of 10k years in age of the genomes has negligible consequences on the estimates).
Split times estimates from G-PhoCS have to be converted into calendar years based on a
mutation rate. Recent work on the high quality genome from Ust’Ishim provides a mutation rate calibrated on ancient DNA, (0.5 × 10−9 per site per year) which is also in line with estimates from high quality modern genomes59. We converted this mutation rate into an appropriate substitution rate for our in vitro deaminated sequence.
|
|

