|
|
Post by Admin on Sept 25, 2021 0:50:24 GMT
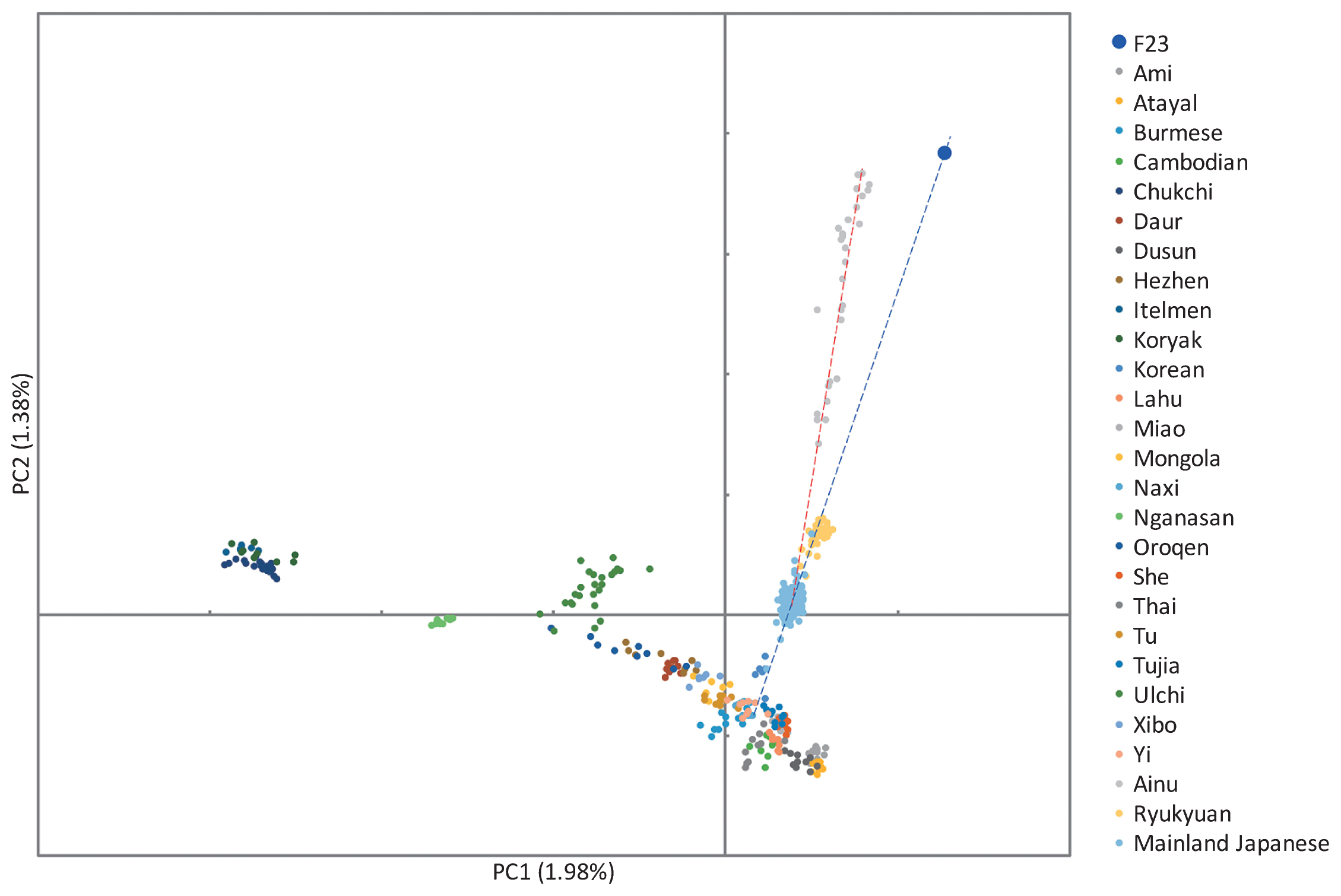 Figure 12 Principal component analysis of F23, Ainu, Ryukyuan, Mainland Japanese, and fully public Affymetrix Human Origins present-day East Asians. Finally, we estimated the proportion of Jomon DNA in three Japanese populations using the f4-ratio-test. When using Korean as a source population, the Jomon ancestry proportion in mainland Japanese was 13%, slightly smaller than the values of TreeMix (15.7%) and qpGraph analyses (9%; Figure 13, Table 5). The proportions were approximately 27% and 66% for Ryukyuan and Ainu, respectively. When more northern populations were used as source populations for testing the Ainu, the Jomon ancestry proportion was decreased. In addition to Jomons, other Northeast Asian populations, such as Ulchi, Hezhen, and Mainland Japanese, as well as Kamchatka people, may have directly or indirectly contributed to the genetic diversity of the Ainu, although we could not identify the source populations in the current study. 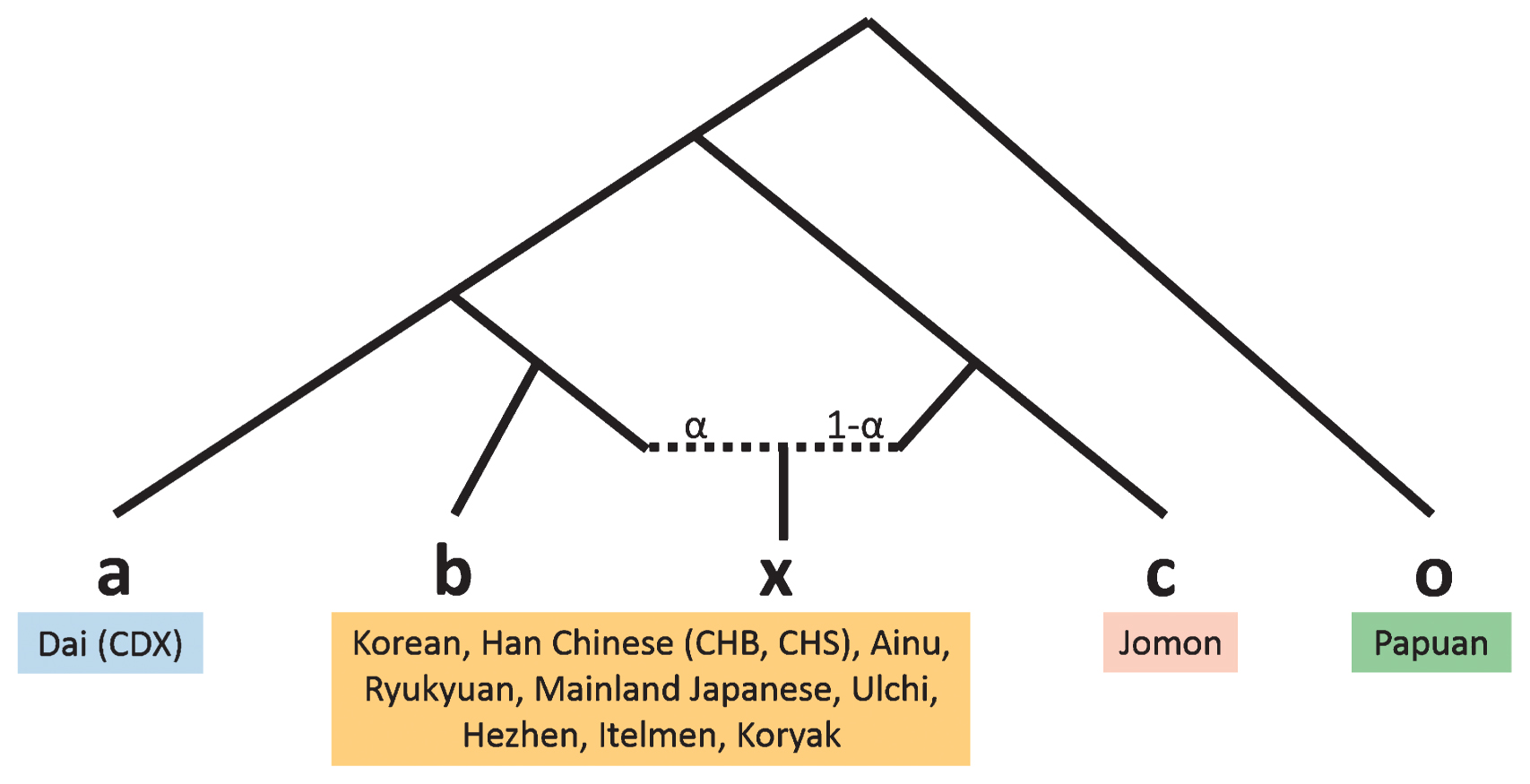 Figure 13 Tree model of the f4 ratio test. o, outgroup; a, population ‘a’, which is related to population ‘b’ but does not contribute to the admixture; b and c, source of the admixture; x, admixed population; α, proportion of admixture. Table 5 Mixture proportions estimated with the ratio f4(CDX, Papuan; Test, F23)/f4(CDX, Papuan; other East Asian, F23) using 130K SNPs a b x c o alpha 1–alpha SE Z-score CDX Korean Mainland Japanese F23 Papuan 0.873 0.127 0.024 36.76 CDX CHB Mainland Japanese F23 Papuan 0.863 0.137 0.015 56.33 CDX CHS Mainland Japanese F23 Papuan 0.816 0.184 0.016 52.48 CDX Korean Ryukyuan F23 Papuan 0.734 0.266 0.024 30.71 CDX CHB Ryukyuan F23 Papuan 0.726 0.274 0.018 39.87 CDX CHS Ryukyuan F23 Papuan 0.686 0.314 0.018 37.63 CDX Korean Ainu F23 Papuan 0.338 0.662 0.040 8.53 CDX CHB Ainu F23 Papuan 0.334 0.666 0.039 8.62 CDX CHS Ainu F23 Papuan 0.316 0.684 0.037 8.57 CDX Ulchi Ainu F23 Papuan 0.509 0.491 0.053 9.57 CDX Hezhen Ainu F23 Papuan 0.457 0.543 0.049 9.29 CDX Itelmen Ainu F23 Papuan 0.869 0.131 0.127 6.87 CDX Koryak Ainu F23 Papuan 0.781 0.219 0.100 7.80 CDX Mainland Japanese Ainu F23 Papuan 0.388 0.612 0.043 9.10 CDX Ryukyuan Ainu F23 Papuan 0.461 0.539 0.048 9.57 |
|
|
|
Post by Admin on Sept 25, 2021 2:07:58 GMT
Discussion This study is the first report of a high-quality genome sequence and Y chromosome haplotype in the Jomon people. Our data provide clear evidence of phenotypic characteristics and phylogenetic relationships among the Jomon, other anatomically modern human populations, and archaic humans. Morphological studies of bones and teeth have shown the unique characteristics of the Jomon people, namely diminutive stature (Suzuki, 1969), smaller teeth than immigrant Yayoi farmers (Brace and Nagai, 1982), prominent glabella, low and broad face with a high nasal bridge (Yamaguchi, 1992), and more frequent non-shovel-shaped teeth than modern East Asians (Matsumura, 1995). Other physiological and morphological traits of Jomon people unidentified from skeletal remains, e.g. alcohol tolerance (Koganebuchi et al., 2017), Rh-blood type, and ear wax (Omoto, 1992), have been inferred indirectly from the geographical distribution of variants in the Japanese archipelago. Recent ancient DNA studies also uncovered phenotypic traits of Jomons, such as ear-wax and ABO blood type (Sato et al., 2009b, 2010). Most of these anthropological traits were observed in F23. One allele of her ABO blood type, Ax02 allele, is rare in modern mainland Japanese (Iwasaki et al., 2000) and other populations; for example, the frequencies of Ax0201 and Ax0202 are 0.006% and 0.015%, respectively, in Germans (Lang et al., 2016). C261del and C297G variants classify her another allele, O-type, into O0201, and the subtype is more frequent in Ryukyuan than in Mainland Japanese (Nakamura, 2008). Importantly, we detected some medically relevant variants relating to disorders. The p.Pro479Leu variant in CPT1A was homozygous in F23. Since this variant was also observed in F5, it is likely that this variant had a high allele frequency in the Funadomari Jomon population. CPT1A is essential for fatty acid metabolism, and the p.Pro479Leu variant in CPT1A is strongly associated with various disorders, such as hypoketotic hypoglycemia and high infant mortality, and with reduced insulin resistance and smaller body size, including weight, lean mass, height, and body mass index (Clemente et al., 2014; Andersen et al., 2016; Skotte et al., 2017). The frequency of this variant is about 56% in Koryak; more than 70% in indigenous arctic populations, including Southwest Alaska Yup’ik, Inuit from Greenland, and Canadian Nunavut Inuit; 90% in Chukchi; and more than 95% in Nunavik Inuit; but is absent elsewhere (Greenberg et al., 2009; Rajakumar et al., 2009; Lemas et al., 2012; Clemente et al., 2014; Zhou et al., 2015; Skotte et al., 2017). Clemente et al. (2014) showed evidence of strong selective sweeps in this variant within the last 6–23 ka in Arctic populations, despite the associated deleterious consequences, and they suggested that this was a result of a selective advantage for either a high-fat diet or a cold environment. Moreover, this amino acid change provided a metabolic advantage for high-fat diet. Hokkaido Jomon people engaged in hunting of not only land animals, such as deer and boars, but also marine fishing and hunting of fur seal, Steller’s sea lions, sea lions, dolphins, salmon, and trout (Nishimoto, 1984). In particular, many relics related to hunting of ocean animals have been excavated from the Funadomari site. A marine reservoir effect was also observed in F23 (77.6%) (Supplementary Table 1). Therefore, it seems reasonable to conclude that they depended on sea animals as a main food source. The presence of the p.Pro479Leu variant seemed to be highly related to their lifestyle. Although Funadomari Jomons shared this rare variant with modern-day arctic populations, suggesting a genetic link between Funadomari Jomons and ancient northern populations, this finding does not necessarily imply that the ancestor of Funadomari Jomons originated from northern East Eurasia since we did not find strong signals from IBD, F-statistics, and fine-STRUCTURE tests. Further studies with more Jomon genomes are needed to determine when and how the variant arose, whether selective sweep occurred independently, and whether this variant was common in (northeast) Jomon people. Our data supported the divergence of the northern Jomon people before the split of the ancestors of Native Americans and East Asians. Moreno-Mayar et al. (2018) estimated that the Native American and East Asian split occurred more than 25000 YBP. Our MSMC results showed that the Funadomari Jomon divergence with Han occurred between 18000 and 38000 YBP. This deep divergence was consistent with the detection of mtDNA haplogroup N9b1 and Y chromosome haplogroup D1b2b. Haplogroups N9b and D1b are localized to the Japanese archipelago, and the ages of the most recent common ancestors of N9b and D1b were estimated to be 22000 (Adachi et al., 2011) and c. 19400 YBP (Hammer et al., 2006), respectively. Evidence of deep divergence supports the idea that northern Jomon people were the descendants of Upper Paleolithic humans in the Japanese archipelago, although it is still possible that Neolithic continental East Eurasians who did not contribute to any present-day East Eurasians may have migrated and become the ancestor of the northern Jomon. Yang and Fu (2018) showed that present-day East Asian populations exhibit remarkable homogeneity relative to ancient samples from other regions and suggested that there was very little diversity in Asia during the Neolithic period, or that this is difficult to find when analyzing only present-day populations. The high genetic similarity between modern populations from the Amur Basin and 7700 year old individuals from Devil’s Gate on the border between Russia and Korea implied that there may have been a high level of genetic continuity in this region during the Neolithic period (Siska et al., 2017), and the distinctive lineage of Neolithic northern Jomon people supports the existence of genetic diversity during the Neolithic period in East Asia. We also found a considerable genetic affinity between Funadomari Jomons and coastal and marine East Asians (Japanese, Ulchi, Korean, Ami, Atayal, and Hezhen). A recent ancient mtDNA study supported the affinity between Ulchi and Hokkaido Jomon populations, who shared mtDNA haplogroups N9b (4.4% and 64.8%), M7a2 (0.6% and 1.9%), and D4h2 (2.5% and 16.7%) (Adachi et al., 2018); among these, N9b and M7a are considered as Jomon genotypes (Adachi et al., 2009). One Jomon genotype, M7a, is also shared with Korean (3.4%) and Philippines (9.4%) (Tanaka et al., 2004). There are two possible explanations for the affinities: (1) sharing the earliest migration into coastal area of East Asia; or (2) gene flow with neighbors after the divergence of the Jomon lineage from other East Asians. The latter is likely in the Japanese and Ulchi because these populations share significantly longer IBD segments with F23 than Korean and other populations. No signals of affinity between Funadomari Jomons and Devil’s Gate, who are likely ancestors of Ulchi, also supported the latter model. However, it is difficult to judge which explanation is the best fit for the history of Korean, Ami, and Atayal in the current data. More genome data from these populations are necessary. In contrast, Funadomari Jomons exhibited less affinity with southeast Asians (e.g. Thai, Cambodian, and Burmese) than for Han Chinese, indicative of the genetic diversity and heterogeneity in Neolithic East Asians. We consider three simple models to explain the relationships between F23, Han, and Southeast Asians (Supplementary Figure 17): (a) unknown populations, who are phylogenetic outgroups of Funadomari Jomon, admixed with the ancestor of Southeast Asians; (b) a gene flow occurred between the ancestor of Funadomari Jomons and an ancient population related to the Han Chinese after the first divergence of the Hokkaido Jomon lineage; and (c) after the ancestor of Funadomari Jomons and continental Northeast Asians split from ancient Southeast Asians, the ancient Southeast Asians strongly admixed with continental Northeast Asians. Pickrell and Pritchard (2012) inferred that Cambodians could trace approximately 16% of their ancestry to a population ancestral to other extant East Asian populations, who are equally related to both Europeans and other East Asians (while the remaining 84% of their ancestry is related to other southeast Asians). If Cambodians are the admixed population between southeast Asians and an unknown Eurasian population, the first model can plausibly explain our results. However, the second and third models may still be valid and should be examined further. In addition, the genetic affinity between Jomons and two Hoabinhians from Pha Faen, Laos, and Gua Cha, Malaysia, is in contrast to these models (McColl et al., 2018). The genetic affinity with Aboriginal Australian in Funadomari Jomons compared with other East Asians also complicates our understanding of the formation of East Eurasians if the affinity is correct. Further analyses of the genomes of continental Neolithic East Eurasians and comparisons among these genomes are necessary to elucidate the detailed structure of the Neolithic population. The deep divergence of the Funadomari Jomon lineage from other populations older than at least 18000 YBP is an indicator of the timing of gene flow between populations. Lazaridis et al. (2014) showed that Finns, Mordovians, Russians (from northwest Russia), Chuvash, and Saami were more related to East Asians than to other Europeans due to East Eurasian gene flow into the ancestors of these northeastern European populations; this event was estimated to occur 1500–2000 years ago. Friedlaender (2005) and Friedlaender et al. (2008) also suggested that Bougainville people admixed with migrants of the more recent Lapita culture 3000 years ago. These estimations are consistent with our results because these populations share more alleles with modern East Asians than with Funadomari Jomons. Yang et al. (2017) showed that 24000 year old MA1 exhibited evidence of gene flow from a population related to East and Southeast Asians. The result of this study supports that the gene flow occurred around or before the split of the Funadomari Jomon lineage. The demographic history of the Northern Jomon people was reconstructed from the genome data in this study. Funadomari Jomons experienced consistently small effective population size after 50000 YBP, with some increases after 25000 YBP. To infer more recent population size changes (<10000 YBP), we need to accumulate more Jomon genomes; however, the high total HBD length in F23 and in southern Native Americans may indicate their small population size during this time. In contrast to southern Native Americans, F23 did not have long HBD fragments, and this and our simulation did not support consanguinity in the Jomon people of Rebun Island. This is consistent with the community structure and network of the Jomon culture. Matsumura et al. (2001) reported that both the morphological similarities and the common custom of tooth ablation suggest biological as well as cultural connections/exchanges between the Jomon people of the northernmost and southwestern regions of Hokkaido. We surmise that the population size changes in the Jomon populations (Koyama, 1978) differed according to region because the Japanese archipelago stretches over 4000 km from north to south and there are various environments. High-quality genomes from southwestern Jomon people will shed light on the relationships between population size changes and different environmental adaptations within the Japanese Archipelago. Previous morphological studies (Matsumura et al., 2001), ancient mtDNA studies (Adachi et al., 2009), and autosomal DNA studies (He et al., 2012; Japanese Archipelago Human Population Genetics Consortium et al., 2012; Kanzawa-Kiriyama et al., 2017) have suggested that all three Japanese populations, i.e. Mainland Japanese, Ainu, and Ryukyuan, inherited Jomon DNA, with the latter two populations retaining more Jomon ancestry. The results of HLA alleles, Y-chromosome haplogroup, and autosomal DNA analysis of Funadomari Jomons are consistent with the previous studies. HLA class I alleles observed in F23 were frequent in the Ainu and Ryukyuan compared with that in the mainland Japanese. Y-chromosome haplogroup D is common in Andamanese (D*), Tibetan (D1a), and Japanese (D1b) populations (Thangaraj et al., 2003) and is rare or absent in other populations. Haplogroup D1b is observed at high frequencies in three Japanese populations: Ainu (87.6%), Mainland Japanese (36.6% and 27.8% in Honshu and Kyusyu, respectively), and Ryukyuans (55.6%) (Tajima et al., 2004); and this Y chromosome haplotype is thought to have contributed to the formation of the Neolithic Jomon males or their ancestral groups in prehistoric Japan (Hammer and Horai, 1995; Tajima et al., 2002). The classification of the F5 Y-haplogroup as D1b2b supports this hypothesis. Intriguingly, 32.7% of Japanese individuals belong to D1b1, whereas only 6.1% belong to D1b* (Naitoh et al., 2013). The frequencies of haplogroup D1b1 and D1b* are consistent with the number of individuals classified into D1b1 (n = 17) and D1b2 (n = 3), which were used in the Y chromosome trees. Because the majority of modern Japanese individuals belong to D1b1, F5 had a rare type. Autosomal DNA analysis also shows the affinities, and the proportions of Jomon DNA in mainland Japanese and Ryukyuan were estimated to be 9–13% and 27%, respectively, which were within the range of ratios reported by Jinam et al. (2015). The current mainland Japanese and Ryukyuan are widely accepted to be a result of admixture of indigenous Jomon and later migrants. The latter were agricultural people who migrated from continental East Asia to the Japanese archipelago probably via the Korean peninsula during and after the Yayoi era. We identified the geographic origins of the agricultural people as populations related to Korean, She, or Han, as previously described by Jinam et al. (2015) and Takeuchi et al. (2017). Additionally, previous studies on nonmetric cranial traits (Shigematsu et al., 2004) and modern/ancient DNA analysis (Tajima et al., 2004; Sato et al., 2007, 2009a, b; Adachi et al., 2018) suggested the considerable genetic influence of the aboriginal people of the Lower Amur/Sea of Okhotsk region on Ainu. In the current study, the Ainu was genetically closest to the Funadomari Jomon, but also received gene flow from ancestral populations that inhabited Kamchatka, Lower Amur River region, and far northeast Siberia. Jeong et al. (2016a) suggested that only the Ainu show a close affinity with the Itel’men, whereas most East Asians are genetically closer to Nganasan from central Siberia, corroborating our f3-statistics results. However, it should be noted that f3-statistics are less sensitive if both source populations are genetically related, e.g. when Ulchi and F23 are used as source populations for Ainu, and it is difficult to recognize Ulchi as a source population. The addition of other ancient genomes (e.g. epi-Jomon, Satsumon, Okhotsk, ancient Sakhalin, and southern Siberian people) and new statistical methods should provide further insights into the genetic history of Ainu. In conclusion, in this study, we characterized a high-quality Jomon genome, which we consider the “reference Jomon genome.” The information presented herein will provide a solid foundation for future genomic studies of additional Jomon individuals excavated from various sites. The addition of new Jomon genome sequences to the current data may provide insights into shared features and/or local adaptations across the Japanese archipelago. In the current study, genome sequences of Sanganji, Ikawazu, and Funadomari Jomon were found to share a common lineage, although the data were not sufficient to identify geographical variations. Based on craniometric data, Kondo et al. (2017) demonstrated that the Jomon people exhibit a discernible level of northeast-to-southwest geographical cline across the Japanese archipelago, placing the Hokkaido and Okinawa Jomons at extreme ends. We hope that more genomic data from various Jomon sites spanning a wider time frame will allow us to elucidate more details of their population history. Supplementary Figures Download (PDF) www.jstage.jst.go.jp/article/ase/127/2/127_190415/_supplement/_download/127_190415_1.pdfSupplementary Tables Download (ZIP) www.jstage.jst.go.jp/article/ase/127/2/127_190415/_supplement/_download/127_190415_2.zip |
|
|
|
Post by Admin on Sept 28, 2021 20:22:21 GMT
Exploring models of human migration to the Japanese archipelago
using genome-wide genetic data
Naoki Osada1*, Yosuke Kawai2
Abstract The origins of people in the Japanese archipelago are of long-standing interest among anthropologists, archeologists, linguists, and historians studying the history of Japan. While the ‘dualstructure’ model proposed by Hanihara in 1991 has been considered the primary working hypothesis for
three decades, recent advances in DNA typing and sequencing technologies provide an unprecedented
amount of present-day and ancient human nuclear genome data, which enable us to refine or extend the
dual-structure model. In this review, we summarize recent genome sequencing efforts of present-day and
ancient people in Asia, mostly focusing on East Asia, and we discuss the possible migration routes and
admixture patterns of Japanese ancestors. We also report on a meta-analysis we performed by compiling
publicly available datasets to clarify the genetic relationships of present-day and ancient Japanese populations with surrounding populations. Because the ancient genetic data from the Japanese archipelago
have not yet been fully analyzed, we have to corroborate models of prehistoric human movement using
not only new genetic data but also linguistic and archeological data to reconstruct a more comprehensive
history of the Japanese people.
Introduction
The origin of the Japanese people—a brief introduction
The Japanese archipelago is at the eastern end of East
Asia, and has a current population of approximately 127
million people. The history of Yaponesians, a term referring
to people prehistorically and historically living in the archipelago
(Yaponesian Genome Project Managing Group,
2020), is of long-standing interest among anthropologists,
archeologists, linguists, and historians studying the history
of Japan. Although many different models for explaining the
origins of Japanese people have been proposed (Nanta,
2008), the ‘dual-structure’ model proposed by Hanihara
(1991) has been considered the primary working hypothesis
for three decades. In the dual-structure model, the present-day
Japanese population consists of two different genetic
layers, which correspond to indigenous Jomon hunter-gatherers and
rice-farming Yayoi migrants. The level of admixture varied across
the archipelago, and the genetic contribution of the Yayoi is weaker
in peripheral regions such as
the Ryukyus (Okinawa) and Hokkaido (represented by
Ainu), where rice farming was introduced at much later
stage than in the central regions of the archipelago (Horai et
al., 1996; Omoto and Saitou, 1997). Hanihara also suggested
that Jomon people entered the Japanese archipelago around
the Upper Paleolithic period and originated from Southeast
Asia, whereas Yayoi people originated from North Asia and
migrated to mainland Japan around 300 BCE. Here, mainland Japan is
defined as Honshu, Shikoku, Kyusyu, and the
surrounding islands (excluding the Ryukyu islands).
Despite its simplicity, later studies largely (but not perfectly)
supported the dual-structure model, but the model
has yet to be corroborated using more newly available data.
For example, recent archeological studies using radiocarbon
dating showed that rice farming was introduced as early as
900 BCE in northern Kyushu, much earlier than previously
thought, then gradually spread to the rest of the archipelago
(de Boer et al., 2020; Leipe et al., 2020). In addition, Saitou
recently proposed an ‘inner dual-structure’ model, which
postulates an additional migration wave to the archipelago,
inferred from the genetic data of present-day and ancient
Japanese (Jinam et al., 2015b; Saitou and Jinam, 2017;
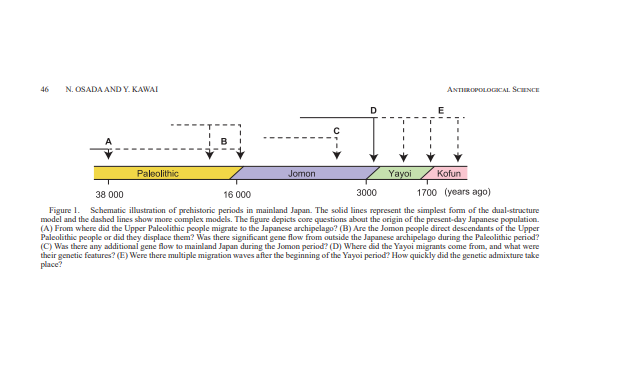
Jinam et al., 2021). In Figure 1, we present the dual-structure
model along with additions related to key questions about
the formation of the present-day Japanese population. The
goal of ongoing research about the issue is to refine or extend
the dual-structure model and to understand how populations in the
archipelago changed genetically.
|
|
|
|
Post by Admin on Sept 28, 2021 22:17:33 GMT
 For over three decades, revealing the history of human populations using genetic data relied mostly on uniparental genetic markers, i.e. mitochondrial genomes and Y chromosomes. However, recent advances in DNA typing and sequencing technologies have provided an unprecedented amount of human nuclear genome data. Because nuclear genomes recombine with each other and contain the information of multiple genealogies, we can obtain information for millions of genealogies from genome-wide genotyping data. The study of nuclear genomes is therefore able to reveal the history of populations on a granular level, even with a relatively small number of samples. In addition, it has become practical to analyze genome-wide polymorphism data of ancient people, even including archaic Homo species such as Neanderthal and Denisovan, providing many findings about prehistoric human migration (Green et al., 2010; Prüfer et al., 2014; Reich et al., 2010). Within just a few years, several key papers analyzed the genome-wide polymorphism data of ancient samples from Southeast, East, and Northeast Asia, including many Paleolithic and Neolithic specimens, and provided numerous novel insights into prehistoric human migration around the Japanese archipelago (Gakuhari et al., 2020; Kanzawa-Kiriyama et al., 2019; McColl et al., 2018; Ning et al., 2020; Wang et al., 2020a, b; Yang et al., 2020). In this review, we concisely summarize recent genome sequencing studies of present-day and ancient people in Asia, mostly focusing on East Asia, and discuss the possible migration routes and admixture patterns of Japanese ancestors. We draw readers’ attention also to recent review papers from different perspectives by de Boar et al. (2020) and Zhang and Fu (2020) for further study. We note that the scenarios presented here are somewhat simplified. The actual migration history of humans would have been much more complex than can be described by a simple narrative. In addition to the concise review, we report on a metaanalysis we performed by compiling publicly available datasets and we clarify the genetic relationships of present-day and ancient Japanese populations with surrounding populations. Because many ancient genome studies have used similar datasets (or reference panels) for their analysis, it is a good opportunity to compare analytical results using different datasets for the validation of previous findings. Here we compile high-coverage whole-genome sequencing (WGS) data of present-day humans and combine them with the variant information of previously published ancient genomes (see Methods). We focus on the analysis of autosomal genome-wide single nucleotide variant (SNV) datasets and do not discuss the details of research using mitochondrial and Y-chromosomal sequences except where necessary. We refer readers to recent papers summarizing the pattern of mitochondrial and Y-chromosomal diversity in Southeast, East, and Northeast Asia for related discussion (Kivisild et al., 2002; Stoneking and Delfin, 2010; Tanaka et al., 2004). |
|
|
|
Post by Admin on Sept 28, 2021 23:13:54 GMT
Paleolithic population history in East Eurasia How anatomically modern humans migrated from Africa to East Asia is a key question for understanding the establishment of present-day Asian populations. Studies of genome diversity in present-day and ancient Asians point to two major routes of prehistoric migration to East Eurasia: the southern and northern routes relative to the Himalayan mountains. Whether the migration through the southern route occurred once or more is under debate both in genetic and in archeological research (Stringer and Andrews, 1988; Macaulay et al., 2005; Malaspinas et al., 2016; Bae et al., 2017; Lipson and Reich, 2017). Along the northern route, it is likely that West and East Eurasian populations were well interconnected during the prehistoric and historic periods via the steppe regions and the Central Siberian plateau (Xu et al., 2008; de Barros Damgaard et al., 2018; Sikora et al., 2019; Wang et al., 2020a). In this section, we summarize how anatomically modern humans migrated out of Africa to East Eurasia and how Paleolithic population movement and admixture formed the current genetic architecture of East and Northeast Asians.  la, the Malay peninsula, and many islands, including Borneo, Java, Sumatra, and the Philippines, formed a landmass called Sundaland, which was connected to the Eurasian continent (Hall and Morley, 2004). Among descendants of the migrants, Papuans, aboriginal Australians, and Philippine Negritos show a larger amount of introgression of genomic fragments with Denisovan ancestry than the other East Asians, including Andamanese (Onge) and Malaysian Negritos (Jehai). This suggests that archaic humans related to the Denisovan lived somewhere in (or nearby) Southeast Asia (Reich et al., 2011; Jinam et al., 2013, 2017). When East Asian populations are clustered as a tree with no assumption of migration between populations, most East and Northeast Asian populations form a single cluster, with Papuans, Australians, and Onge people at root positions, suggesting that the Southern route predominantly shaped the genetic features of present-day East and Northeast Asian populations (HUGO Pan-Asian SNP Consortium, 2009; Mallick et al., 2016; Lipson and Reich, 2017;). |
|

