|
|
Post by Admin on Nov 5, 2016 21:01:30 GMT
 Figure 1. The key statistic used to support the claim of unknown archaic ancestry in Andamanese and mainland Indians by Mondal et al. is inconsistent with all previously published datasets. Genomic studies have documented a contribution of archaic Neanderthals and Denisovans to non-Africans 1,2. Recently Mondal et al.3 published a major dataset—the largest whole genome sequencing study of diverse South Asians to date—including 60 mainland groups and 10 indigenous Andamanese. They reported analyses claiming that nearly all South Asians harbor ancestry from an unknown archaic human population that is neither Neanderthal nor Denisovan. However, the statistics cited in support of this conclusion do not replicate in other data sets, and in fact contradict the conclusion. The main evidence cited by Mondal et al. is statistics suggesting that indigenous Andamanese and mainland Indian groups share fewer alleles with sub-Saharan Africans than they do with Europeans and East Asians; such statistics have previously been reported in Australasians, for whom they represent key evidence of Denisovan admixture2. To document their signal, Mondal et al. compute D-statistics1,4 of the form:D(Ancestral allele, African; East Asian, X) (Equation 1)  These statistics test the hypothesis of an equal rate of allele sharing of East Asians and X with Africans. In Mondal et al.’s computation, these statistics are negative when X is any Indian group or Andamanese, a result they interpret as evidence of more archaic ancestry than in East Asians. As they find no evidence of excess allele sharing with Neanderthals or Denisovans, they argue that the contribution is from an unsampled archaic lineage. We sought to replicate these statistics in two single nucleotide polymorphism (SNP) data sets of ~600 thousand SNPs each5,6,4,7, whole genome sequence data from the 1000 Genomes Project phase 3 (~78 million SNPs)8, and high-coverage genomes (~34 million SNPs)9. There is no evidence for excess archaic ancestry in South Asians in any of these four data sets (Figure 1), and in fact the values reported by Mondal et al. (D =-0.024 ±0.004; Supplementary Table 13 of their study3) are inconsistent with those in each of these other datasets (all P < 10-5 by a one-tailed test).  Mondal et al. also report statistics suggesting more archaic ancestry in indigenous Australians than in indigenous Papuans, as reflected in D-statistics that are far more skewed from zero when X=Australian than when X=Papuan3. They interpret this as evidence of unknown archaic ancestry in Australians to a greater extent than in Papuans. However, we do not replicate this excess when recomputing this statistic using highcoverage genomes from these populations: D(chimpanzee, Yoruba; Dai, Australian) = -0.031 ± 0.003 and D(chimpanzee, Yoruba; Dai, Papuan) = -0.029 ± 0.003. In addition, a direct comparison between Australians and Papuans provides no evidence for a difference: D(chimpanzee, Yoruba; Australian, Papuan) is only Z=0.6 standard errors from zero9,11. These findings support the notion that Papuans and Australians descend from a homogeneous ancestral population, and are inconsistent with the suggestion that Australians harbor much more archaic ancestry than Papuans.  In fact, some of the statistics computed by Mondal et al. directly contradict their proposed model of unknown archaic ancestry specific to Indians and Andamanese. Figure 1b of Mondal et al. suggest that the Riang (RIA)—a Tibeto-Burman speaking group from the northeast of India for which sequencing data are newly reported in the study—derive almost all of their ancestry from the same East Asian lineages as populations like Dai and Han Chinese, which in Figure 1b of Mondal et al. have no evidence of unknown archaic ancestry. Under the authors’ hypothesis of more archaic ancestry in lineages that are unique to South Asia than in lineages shared with East Asians, one would not expect a significant statistic in the Riang, but in fact the signal is just as strong as it is for the Andamanese Onge, Andamanese Jarawa, mainland Irula and mainland Birhor, the great majority of whose ancestry is inferred to derive from lineages unique to South Asia. One possible explanation for the skew that the authors observe3 is batch artifacts, reflecting differences in laboratory or computer processing between the data newly reported by Mondal et al., and the data from non-Indians used for comparison10. Separate processing is known to be able to cause correlation of errors within datasets, and this could explain why the newly reported South Asian genomes appear (artifactually) to share fewer alleles with other modern humans. However, the data used by Mondal et al. have not been made available for independent reanalysis, and without this, a definitive explanation is not possible. Whatever the explanation, our analyses contradict the claim of unknown archaic ancestry in South Asians. |
|
|
|
Post by Admin on Dec 12, 2016 20:34:46 GMT
 The Onge people exclusively belong to mtDNA haplogroup M and Y-DNA Haplogroup D, which is quite similar to the Tibetans and the Ainu in East Asia, but Malaysian Negrito subgroups are genetically distant from East Asian populations. The Malaysian Negritos are much closer to the Onge of the Andaman Islanders and a recent study by Aghakhanian et al. (2015) found significant gene flow between Andamanese and Malaysian Negritos. There’s also substantial Austro-Asiatic admixture in these Malaysian Negrito subgroups and the Onge and Jarawa appeared closest to Papuans and Melanesians from PCA results. To understand population structure across Negritos, other OA subgroups, and their relationship with neighboring populations in Asia and Oceania, a PCA was performed (fig. 2 and supplementary fig. S1, Supplementary Material online). As presented in figure 2A, the first component, which captures 32% of total variation, clearly distinguishes South Asian populations from those in the East. From PC2, the Onge and Jarawa, both Negrito subgroups, clustered together and were distinct from other populations. However, they appeared closest to Papuans and Melanesians. The Malaysian Negrito subgroups, while clustering closer to East Asians, showed a tendency toward other Negrito subgroups in Oceania and Andaman islands. The rest of OAs such as Senoi and Proto-Malays as well as Singaporean Malays were located between Malaysian Negritos and East Asian clusters indicating that these groups might be admixed between these two populations. However, both Senoi and Proto-Malay groups lay closer to East Asians on PC4 suggesting that all these populations may have a common origin.  FIG. 2.— PCA of Orang Aslis and surrounding populations. Like PCA analysis, the results of Neighbor-net tree showed that OAs are closest to EA populations. As evident in supplementary figure S2, Supplementary Material online, all four subgroups of Negritos formed a clade, while Senoi and Proto-Malay were positioned at various points between these two clades. The long branches observed in Bateq, Jehai, Kintaq, CheWong, Seletar, and MahMeri suggest strong drift in each of these populations. Interestingly, Seletar located between Malaysian Negritos and Oceanians. The tree also indicated genetic affinity between Andamanese and Oceanians. In order to determine critical ancestral components that may have shaped the genetic architecture among the OAs, we applied ADMIXTURE analysis. The results of ADMIXTURE from K = 2 to K = 12 are shown in figure 3. Each individual is represented as a vertical bar and their corresponding ancestry components are shown by different colors. Different colors indicate different ancestry lineages. As presented, K = 2 separated Central-South Asia (red) and EA (yellow) and the latter appears to be the major component in all OA groups. From K = 3, Andamanese component (pink) appeared. This component also presented considerably in Oceanians and in lesser extent in Malaysian Negritos. At higher K = 4 and K = 5, Negrito (dark green) and Oceanian (dark blue) components appeared respectively. The best model which had the lowest cross validation error suggests nine major ancestral groups which gave rise to the 40 distinct populations included in our study. At K = 9, all Negrito subgroups showed similar ancestral patterns. However, we observed small portions of other ancestral components (shown in yellow and purple) in some Negrito individuals (especially Mendriqs).  FIG. 3.— ADMIXTURE analysis of Orang Asli, Andamanese, South Asian, and East Asian ethnic groups from HGDP and Singaporean Malay. To understand the relationship between our populations and examine the gene flow between them, we used TreeMix (fig. 4 and supplementary fig. S3, Supplementary Material online). Using Yoruba as root, the graph that best fits our data (99.4% of variances) inferred six migration events. The tree topology was consistent with geographical distribution of populations and with previously shown Neighbor-net tree. Andamanese and Oceanians grouped together in a deep clade, while all OA groups formed a distinct cluster. Focusing on migration events, a migration (migration weight 0.37) directed from root Onge and Jarawa toward Malaysian Negrito root. The resulting tree also highlighted another migration (0.39) from the root of Bateq and Jehai to CheWong. To further investigate gene flow between OAs and other populations, we used D statistics (table 1 and supplementary tables S2 and S3, Supplementary Material online). The computed D statistics demonstrated significant gene flow between Andamanese and Malaysian Negritos but there was no significant gene flow detected between Andamanese and other OA groups. This suggests that an earlier gene flow occurred before other OA groups arrived in Peninsular Malaysia. The D statistics supported admixture between different OA groups, as gene flows between Negrito/Senoi, Negrito/ Proto-Malays, and Senoi/Proto-Malays were evident. We also traced admixture in AA-speaking OAs and those of Mainland SEA and Lahu and Dai, ethnic groups from South China.  FIG. 4.— Treemix tree of Orang Asli subgroups, Negrito groups of Andaman Islands, and South and East Asian populations from HGDP. On a finer scale, Malaysia Negrito subgroups were clearly different from EA populations. This distinct pattern may have resulted from genetic drift. It is also conceivable that they had longer periods of isolation from other inhabitants in the region, as indicated by Fst and LD decay. The ancestral component (dark green) “belonging” to Malaysian Negritos was also spread among Southeast Asian and Southern Chinese populations. However, although Negritos predominantly shared this ancestral component, the Mendriq shared more portions of other ancestral components with East Asians and Senoi. This suggests more recent gene flow between them and their neighboring populations, most likely Malays. A similar observation was reported in Jehai, a Negrito subgroup using a less SNP (Jinam et al. 2013). The Senoi and Proto-Malay were closely related to EA, either because they share relatively recent common ancestors or because of recent gene flow. However, different patterns emerged in Seletar and CheWong. The corresponding ancestral component of Seletar, a subgroup of Proto-Malay, emerged at K = 6 in ADMIXTURE and Neighbor-net tree showed an affinity to the Oceanian. Anthropological information regarding origins of the Seletar is scarce and anecdotal. There is a paucity of information about this community. It is plausible that Seletar might have experienced a recent bottleneck as suggested by the long stretches of LD in their genome. The low levels of mtDNA diversity (Jinam et al. 2012) also provide support for the likelihood of a bottleneck in this population. ADMIXTURE and TreeMix results from CheWong suggest that they are intermediate between Negritos and Senois. Because CheWong appeared distinct at K = 11, it can be inferred that their ancestors experienced one or possibly more admixture events in the past, and later became isolated from founding populations. The argument for CheWongs to be admixed is supported by several factors. First, the cultural practices of CheWong are more similar to other Senoi rather than Negritos, while their language is northern Aslian, similar Negrito dialects. Physically, they appear to have intermediate phenotypes between Negrito and Senoi. The genetic evidence presented here for the first time may reduce disagreement among various anthropologists who study tribes in SEA (Benjamin 2013). Genome Biol Evol (2015) 7 (5): 1206-1215. doi: 10.1093/gbe/evv065 |
|
|
|
Post by Admin on Mar 17, 2017 20:20:33 GMT
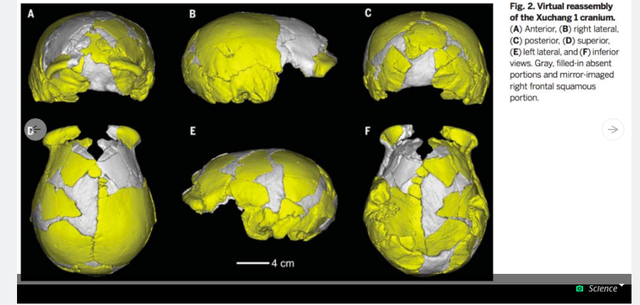 Zhan-Yang Li, Wu's colleague at the Institute for Vertebrate Paleontology and Paleoanthropology (IVPP) in Beijing, found miraculously undamaged fragments of the two crania in Lingjing, a village in Henan, China. A spring flowed there during the Pleistocene period when these humans would have lived, and the area was full of now-extinct megafauna like Bos (aurochs, or wild cows), Megaloceros (a massive deer), and Coelodonta (a rhino), as well as elk and horses. Bones from these animals were found with Xuchang 1 and 2, along with stone tools made from quartz. It appears that Xuchang 1 and 2 were successful hunters with a rich array of foods to eat. They were also part of a regional group of "new or unknown archaic humans" never before seen by paleoarchaeologists in the West. Their unique "mosaic" of modern and Neanderthal traits, say Xu and colleagues in their paper, is "not known among early Late Pleistocene humans in the western Old World." It also suggests that these people came mixed with Neanderthals and other ancient populations, possibly a few times over thousands of years. 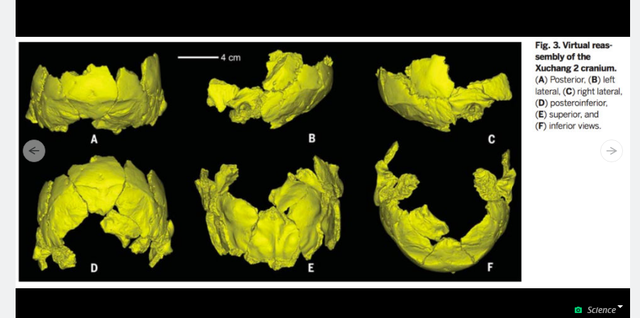 Speaking with Science News, University College London anthropologist María Martinón-Torres said one possibility is that these are the first crania discovered from Denisovans, "something with an Asian flavor but closely related to Neandertals." Denisovans are an archaic human whose DNA was sequenced based on a few finger bones and teeth. Like Xuchang 1 and 2, they combined traits of modern humans and Neanderthals. But anthropologist Erik Trinkaus, a co-author on the new study, said he didn't want to use the term Denisovan because "it's a DNA sequence" and nothing more. Trinkaus was one of the first scientists to popularize the idea that modern humans and Neanderthals had children together. He based this hypothesis on the shape of several early human fossil skulls, and DNA analysis of modern humans eventually validated his claims. We now know that many humans alive today, especially those from Europe and Asia, have traces of Neanderthal DNA in their genomes. What's perhaps most interesting about Xuchang 1 and 2 is their place in the grand sweep of human evolutionary history. Wu and her colleagues call them "critical" to understanding how modern humans spread throughout the Old World. They note that modern human biology was establishing itself via migrations and mixes for hundreds of thousands of years throughout many regions in Africa, the Old World, and southeast Asia. Given the nature of our evolution during the Pleistocene, it's likely that mosaic humans like Xuchang 1 and 2 might have been the rule, not the exception. |
|
|
|
Post by Admin on Mar 18, 2017 20:07:29 GMT
 Fig. 1. Modern human genetic variation projected on axes of variation defined by chimpanzee, Denisova, and Neandertal. (A) A model of hominin evolutionary history suggested in the work in ref. 24 with putative admixture events indicated by arrows. (B) Population means for each of 62 populations. (C) PC1 in individuals from Eurasia and America as a function of distance from East Africa (kilometers). (D) Interpolated spatial distribution of PC2 [transformed for visualization as (−1) × log10 (x + C), where x is the PC loading and C = 0.04231] in individuals from Eurasia, Oceania, and America. (E) Interpolated spatial distribution of the frequency of Denisova alleles at SNPs where Denisova is different from chimpanzee and Neandertal. Sample localities are indicated with rectangles. In this study, we analyzed patterns of genetic variation in modern humans in the light of the two archaic genomes using genotype data from a diverse set of extant populations, and we found a signal of Denisova admixture in contemporary East Asian populations. We also studied the effect of ascertainment bias under serial founder models of human expansion to show that the signal of Denisova admixture in contemporary East Asians is opposite to the expectation under a model of solely Neandertal admixture with the ancestral population of non-Africans followed by greater genetic drift in East Asia than in Europe (40). Principal Component Analysis of Archaic Ancestry. We performed principal component analysis (PCA) (44) by defining the first two principal components (PCs) using the Denisova, the Neandertal, and the chimpanzee and projected extant humans on the resulting axes of variation (24). This setup resulted in PC1 describing general genetic similarity to archaic humans (represented by both the Neandertal and Denisova genomes) and PC2 contrasting genetic similarity between Neandertal and Denisova. Under the assumption of a common shared history between Neandertal and Denisova (24) as well as no admixture between archaic populations and the ancestors of extant human populations since the diversification of modern humans (Fig. 1A), extant individuals are expected to be homogeneously distributed between archaic human and chimpanzee variations (24) (SI Materials and Methods).  Fig. 2. PCA of archaic ancestry under a serial founder model with (or without) archaic admixture. PCA was performed by projecting hypothetical extant humans onto the variation of a hypothetical chimpanzee and two hypothetical archaic hominins that shared a recent history with modern humans ∼400 kya. (A) A model with no admixture between colonizing and archaic populations. (B) A model with two separate admixture events: from archaic population A into colony 25 (forward in time, 2.5% of colony 25 is replaced) and from archaic population B into colony 97 (forward in time, 5% of colony 97 is replaced). This model is similar to the model suggested in ref. 24 (Fig. 1A). (C) A model with the two admixture events in B and an additional admixture event from archaic population B into colony 75 (forward in time, 1% of colony 75 is replaced). D–F show PCA results from models A, B, and C, respectively. G–I show PCA results for models A, B and C, respectively, but with SNPs with minor allele frequency (MAF) < 5% excluded. In D–I, we display the mean PC loading of 10 samples from each colony numbered and colored according to distance from the founding population. The model in C produced the qualitatively most similar result compared with the empirical data in Fig. 1B. We recovered the previously reported (24) pattern—that extant human variation is largely organized in three clusters corresponding to Africans, Oceanians, and other non-Africans, respectively (Fig. 1B and Fig. S1). In the worldwide collection of extant populations, we used Procrustes superimposition (45) to compare PC1 and PC2 with geographic coordinates (longitude and latitude) for each population and found that sampling location was mirrored, to some extent, by archaic ancestry (individuals: Procrustes correlation = 0.127, P << 10−6; population means: Procrustes correlation = 0.309, P < 0.01). This pattern was expected given a model with two episodes of archaic gene flow involving non-Africans and Oceanians (Fig. 2B) (23, 24). However, PC1 and PC2 were also correlated with geography in a region comprising Eurasia and the Americas (individuals: Procrustes correlation = 0.104, P < 10−4; population means: Procrustes correlation = 0.335, P = 0.017), which seemed incompatible with previously suggested admixture scenarios postulating that archaic ancestry is homogeneous in people of European, Asian, and Native American descent (23, 24). To disentangle the signal of Denisova admixture from the signal of Neanderthal admixture, we investigated the distribution of PC2, which separated Denisovans and Neandertals, and found that proximity to Neandertal increases with distance from Africa (individuals: rs = 0.078, P = 0.010). However, East Asians were an exception to this trend in that they were significantly closer to Denisovans compared with Europeans (P = 0.003), all West/Central Eurasians (P = 0.006), and Americans (P < 10−4). In a spatial interpolation of log-transformed PC2 values, the affinity to the Denisova genome seemed to be strongest in Southern China and Southeast Asia (Fig. 1D), which is noteworthy considering the supposed East Eurasian distribution of Denisovans (24). To investigate this geographical pattern using an alternative approach, we identified SNPs in chimpanzees and Neandertals that shared the ancestral allele, and Denisova had the derived allele; we computed the frequency of the derived Denisova allele in global modern human populations. Using the same method of spatial interpolation, we found that East Asian populations, particularly Southeast Asian populations, had, on average, a greater frequency of the derived Denisova allele compared with other populations (except for Oceanians) (Fig. 1E). For example, although the greatest Denisova allele frequency was found in Papuans (53.5%), Yizu from Southern China had a greater frequency of the Denisova allele than Melanesians from Bougainville (53.0% vs. 52.9%) (Table S3). |
|
|
|
Post by Admin on Mar 19, 2017 20:03:13 GMT
Formal Test for Archaic Admixture. To test the hypothesis of Denisovan ancestry in East Asians using an alternative approach, we performed 4-population tests (23, 24, 40, 47, 48) on diploid genotype data from different regions. Note that using population allele frequencies is confounded by demographic effects (48) but may also provide more power and a more diverse set of samples compared with using low-coverage shotgun sequences from a smaller set of individuals (23, 24). To investigate fine-scale geographical patterns suggested by the PC analyses (Fig. 1 D and E), we divided the East Asian populations in our dataset into two groups, South and North of Beijing (with the North subgroup including Beijing and Japan). We then tested the hypothesis of no gene flow in the population topology (Denisova, chimpanzee), (Northeast Asians, Southeast Asians) and found that allele frequencies in the Southeastern Asian group were significantly more similar to the Denisova data (D = 0.55 ± 0.23% SE, Z = 2.40) (Table S4). In addition, we found that allele frequencies in the Southeastern Asian group were significantly more similar to the Denisova genome compared with the Neandertal genome (D = 0.66 ± 0.30%, Z = 2.22). This differentiation is not predicted by the effects of ascertainment bias, which was observed in our simulations, because ascertainment bias is expected to artificially increase similarity to Neandertal under the model suggested by previous studies (23, 24). Indeed, the only other pairwise regional comparisons for which we see a strong pattern of similarity to Denisova compared with both chimpanzee and Neandertal include Oceanians (Fig. 3 and Table S4).  Fig. 3. Results of 4-population tests suggest Denisova-related ancestry in Southeast (SE) Asia. Z scores for the D statistic in all pairwise comparisons between Africa, Middle East, Central/South Asia, Southeast Asia, Northeast (NE) Asia, Oceania, and America are displayed. The configuration of each pairwise comparison that gives a positive value of D in the test (Pop1, Pop2, Denisova, chimpanzee) was chosen to ease visualization. Except for Oceanians and the comparison between SE Asia and NE Asia, populations that show high affinity to Denisova compared with chimpanzee tend to also show a higher affinity to Neandertal compared with Denisova (negative values on the y axis). Comparisons with Africans are shown by triangles. The area corresponding to significant deviations from 0 (|Z| > 2) is shaded, with the overlap representing significant deviations for both tests (Table S4). The reason why an affinity to Denisova is not as clearly seen in 4-population tests between Southeast Asians and other populations (except Northeast Asians) (Fig. 3 and Table S4) could be the joint effect of ascertainment bias and genetic drift, which is indicated by our simulations. For example, a test of the topology [archaic population A, archaic population B, (colonies 50–70), (colonies 97–100)] fails to detect a fraction of 5% ancestry contributed by archaic population B to colonies 97–100 in simulation model ii (Fig. 2B), with filtering for minor allele frequency > 5%, and in fact, it suggests a skew toward colonies 97–100 being more similar to archaic population A (D = 1.0 ± 3.7%, Z = 0.27). In general, a signal of archaic ancestry is detected only if tests are performed between populations in which the magnitude of genetic drift has been similar. For example, if colonies 50–70 are substituted with colonies 90–96 in the test above, the test statistic significantly deviates from zero, and the admixture is detected (D = −7.0 ± 2.7%, Z = −2.6), although colonies 50–70 have exactly the same true fraction of archaic ancestry as colonies 90–96. However, the admixture is detected in both cases (Z < −8) if the data are unascertained (no minor allele frequency filtering). Although this Denisova ancestry in Southeast Asians could possibly have been introduced by gene flow from indigenous Oceanian populations after the introduction of Denisova-related genetic variants in Oceanians, evidence for such large-scale migration has not been found (56, 57). Moreover, this gene flow would have had to be substantial enough not to dilute the Denisova ancestry present in Oceanians beyond detection. Assuming ∼5% Denisova-related ancestry in Oceanians (24), any fraction of Denisova-related ancestry in Southeast Asia would require a ∼20 times as large contribution from Oceanian populations to be explained solely by modern human gene flow. Quantitative estimation of the precise fraction of Denisova-related ancestry in Southeast Asian populations based on genotype data are unfortunately sensitive to ascertainment bias and genetic drift, and such estimates will require genome sequence data that are currently unavailable. However, both the PCA results (Fig. 1B) and the approximately six times lower absolute values of the D statistic in tests between Northeast Asians and Southeast Asians compared with tests between Northeast Asians and Oceanians (Table S4) indicate a relatively low fraction of Denisova-related ancestry. Thus, the fraction is likely to be smaller than both the ∼5% fraction of Denisova-related ancestry present in Oceanians and the ∼2.5% fraction of Neandertal ancestry present in non-Africans (23, 24), perhaps around 1%. The lack of evidence of Denisova ancestry in other Eurasian populations indicates that this genetic material was introduced into the ancestral population of Southeast Asians after the time of divergence from Europeans, a date that has been estimated to 23–45 kya (58, 59) but could also have occurred considerably earlier (27, 28, 30). The apparent absence of Denisova ancestry in Native Americans in our study could be influenced by the biased affinity to the Neandertal genome that is expected because of ascertainment bias and genetic drift, but analyses of unascertained low-coverage shotgun sequence data from a single Native American individual resulted in a similar conclusion (24). If absent in America, the Denisova component must have appeared in the ancestors of East Asians after their divergence from Native Americans, which has been dated to ∼14–30 kya (58, 60, 61). PNAS November 8, 2011 vol. 108 no. 45 18301-18306 |
|

