|
|
Post by Admin on Sept 1, 2018 18:48:34 GMT
 Differences in DNA sequences correspond to nucleotide substitutions that have accumulated since their split from a most recent common ancestor (MRCA). When the average number of substitutions occurring per unit of time can be determined, the “molecular clock” rate can be estimated. Under the assumption of constant rates of change among lineages, molecular clocks have been used to estimate divergence times between closely related species or between populations. Fossil evidence has been frequently used to estimate a date for the MRCA of two related groups, thus providing a calibration point for the molecular clock. The sparseness of the fossil record, however, poses limitations on the reliability of such estimates. For example, in human evolution, no fossil has yet been identified to represent the uncontested MRCA for humans and chimpanzees or other closely related primate species. As a consequence, the nuclear and mitochondrial mutation rates for the human lineage have been heavily debated [1]. Recent analyses of de novo substitutions from genome sequencing of parent and offspring trios allow the direct calculation of nuclear substitution rates per generation. This alternative to the fossil calibration of the human molecular clock is arguably more accurate. Surprisingly, publications using this approach have recently pointed to de novo rates that are about half the value of those previously determined from fossil calibrations [1, 2, 3, 4, 5]. A slower substitution rate has important implications for inferring the timing of key events in human evolution such as our divergence from our common ancestor with chimpanzees, our divergence from Neanderthals and Denisovans, and the migration of modern humans from one region or habitat to another. Taking these new rates into consideration, most date estimates would be pushed back by a factor of two—for example, yielding a West African/non-African split date of 90–130 kya, which is up to 60 kya older than some previous estimates [1].  Attempts at calculating the human mitochondrial DNA (mtDNA) substitution rate have relied on either estimates derived from fossil calibration or archaeological evidence of founding migrations; however, reliance on a single calibration date can easily lead to a biased rate estimate [6]. Estimates of substitution rates for the coding region of the mtDNA have ranged from 1.26 × 10−8 substitutions per site per year [7], calibrated with a chimpanzee-human divergence time of 6.5 million years before present (BP), to 1.69 × 10−8, inferred from an accepted date of 45 kya for the peopling of Oceania [8]. This 45 kya date is based on radiocarbon-dated archaeological material, thus providing a calibrated age for the haplogroup (hg) Q lineage that is unique to contemporary populations from this region. Soares et al. [9] considered both the coding and noncoding regions in their estimate to accommodate the effects of natural selection and arrived at a rate of 1.67 × 10−8 substitutions per site per year using a calibration point of 7 million years BP for the chimpanzee-human split.  An alternative approach for obtaining greater precision in measuring substitution rates is through the analysis of genetic data from ancient samples for which reliable radiocarbon dates are available. Ancient humans are well suited to provide calibration points for the human mitochondrial molecular clock: reliable radiocarbon dates are available for many specimens; hence, the number of substitutions that have accumulated among lineages can be directly translated into the number of substitutions per site per year. Branch shortening—the effect of fewer nucleotide substitutions on ancient branches in a phylogenetic tree as compared to modern—is commonly observed in phylogenetic studies of ancient humans [10]. The observed branch shortening reflects the comparatively shorter time since the common ancestor for the ancient human as compared to the present-day individual: a present-day lineage has had more time to accumulate nucleotide changes. If we know the age of the ancient sample (e.g., from a carbon date), we can thus infer the mutation rate necessary to produce the observed degree of branch shortening. Here we use the complete or nearly complete mitochondrial genomes from ten ancient modern humans for which reliable radiocarbon dates are available to calculate the human mtDNA substitution rate directly. This strategy circumvents the limitations imposed by the use of indirect measures of substitution rates such as those obtained via fossil calibration. The samples used in this analysis span 40,000 years of human history and originate from Europe and Eastern Asia. We use our substitution rate to estimate the dates of major human evolutionary events in the last 200,000 years. Of particular note, our rate suggests an upper bound on the split between non-Africans and sub-Saharan Africans of less than 95 kya. Even though this estimate is a conservative upper bound because the genetic divergence at any particular locus in the genome is by definition older than the population divergence time, this date, is on the extreme lower end of population split times estimated from de novo substitution rates for nuclear DNA [1]. |
|
|
|
Post by Admin on Sept 2, 2018 19:11:58 GMT
Evolutionary Analysis All but one of the ancient modern human sequences from Europe belonged to mtDNA hg U, thus confirming previous findings that hg U was the dominant type of mtDNA before the spread of agriculture into Europe [15]. The exception was the Cro-Magnon 1 sample, which belonged to the derived hg T2b1, an unexpected hg given its putative age of 30,000 years [16]. Since the radiocarbon date for this specimen was obtained from an associated shell [16], we dated the sample itself using accelerator mass spectrometry (AMS). Surprisingly, the sample had a much younger age of about 700 years, suggesting a medieval origin. Consequently, this bone fragment has now been removed from the Cro-Magnon collection at the Musée de l’Homme in Paris. Attempts to directly date other remains from the Cro-Magnon type collection unfortunately failed. The good molecular preservation of our sample for both DNA and AMS dating, in contrast, suggests that this particular bone has a different origin from the other remains in the collection. For the remaining eight ancient Europeans, we built a phylogenetic tree for hg U, which included 63 contemporary mtDNAs from this haplogroup. The tree clearly shows that all four Paleolithic pre-Last Glacial Maximum (LGM) samples display a short branch compared to the four ancient post-LGM samples (Figure S2). Predictably, the older samples Dolni Vestonice 14 and 15 fall in a basal position relative to the contemporary mtDNA hg U5. The Tianyuan sequence from Eastern China falls basal to the contemporary hg B, common in most parts of Eastern Asia, Oceania, and the Americas. The mtDNA genetic diversity that we measure in early modern Europeans is about 2-fold less than the mtDNA diversity in today’s Europeans, but about 1.5 times higher than that measured in Neanderthals contemporary with these early modern humans [17] (excluding the older Mezmaiskaya individual) (Table S3). Although these measurements provisionally suggest that a higher population size might have contributed to early modern humans outcompeting Neanderthals after their arrival in Europe, there are caveats to this analysis. First, we have a limited sample size of ancient specimens. Second, we have sampled from several different time periods, a practice that overestimates actual genetic diversity [17]. Third, our sampling is nonrandom; for example, we included several individuals from within the same burial site (Dolni Vestonice), where maternal relatedness is possible and would give an underestimate of true diversity. More data are necessary to provide a definitive assessment of the genetic diversity of these prehistoric populations. Haplogroup Divergence Time Estimates Using the substitution rate for the whole mtDNA genome obtained by Bayesian estimation, we estimated the time of the MRCA for all modern humans at 157 kya (120–197 kya, 95% HPD) (Table 2). Our rate also implies a split of all non-African hgs from the closest widespread sub-Saharan African hg (L3) of 78.3 kya (62.4–94.9 kya) (Table 3). The MRCA of hg Q, often referred to as a maximum age for the settlement of Australia, was calculated at 42 kya (30–54.9 kya) (Table 3). The time to most recent common ancestor (TMRCA) of hg U5, often argued to have evolved within the first early modern humans in Europe [18], was calculated at 29.6 kya (22.7–37.2 kya) (Table 3). 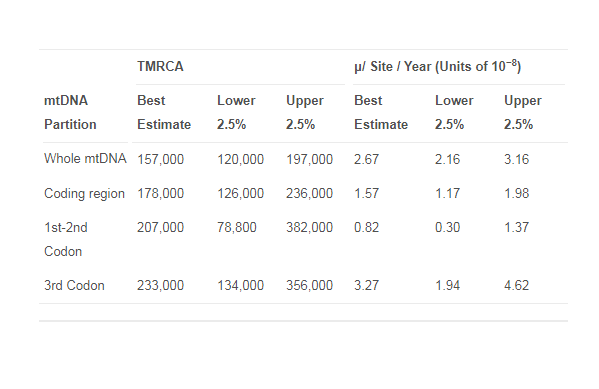 Table 3. TMRCA of Different Haplogroups To test whether the inferred mutation rates are dependent on a single directly dated mtDNA sequence (which in principle could have an inaccurate carbon date), we carried out a Bayesian MCMC analysis for the coding regions with a constant size model and relaxed clock using each mtDNA sequence older than 4,000 years independently as separate tip calibrations. The results range from 1.14 × 10−8 substitutions per site per year for the Kostenki specimen to 4.5 × 10−8 substitutions per site per year for the Boshan specimen (Table S1). The confidence intervals for all samples overlap the value obtained for the whole data set, suggesting that no single sample is driving our overall mutation rate estimate. |
|
|
|
Post by Admin on Sept 4, 2018 19:35:26 GMT
Discussion We were able to reconstruct three complete and six nearly complete mitochondrial genomes from ancient human remains that were found in Europe and Eastern Asia and span 40,000 years of human history. All Paleolithic and Mesolithic European samples belong to mtDNA hg U, as was previously suggested for pre-Neolithic Europeans [15]. Two of the three individuals from the Dolni Vestonice triple burial associated with the pre-ice age Gravettian culture, namely, 14 and 15, show identical mtDNAs, suggesting a maternal relationship. Furthermore, both individuals display a mitochondrial sequence that falls basal in a phylogenetic tree compared to the post-ice age hunter-gatherer samples from Italy and central Europe, as well as the contemporary mtDNA hg U5 (Figure 1). It has been argued that hg U5 is the most ancient subhaplogroup of the U lineage, originating among the first early modern humans in Europe [18]. Our results support this hypothesis because we find that the two Dolni Vestonice individuals radiocarbon dated to 31.5 kya carry a type of mtDNA that is as yet uncharacterized, sits close to the root of hg U, and carries two mutations that are specific to hg U5. With our recalibrated molecular clock, we date the age of the U5 branch to approximately 30 kya, thus predating the LGM. Because the majority of late Paleolithic and Mesolithic mtDNAs analyzed to date fall on one of the branches of U5 (see also [15]), our data provide some support for maternal genetic continuity between the pre- and post-ice age European hunter-gatherers from the time of first settlement to the onset of the Neolithic. U4, another hg commonly found in Mesolithic hunter-gatherers [15], has so far not been sequenced in a Paleolithic individual, and we find hgs U8 and U2 in pre-LGM individuals but not in later hunter-gatherers. At present, the genetic data on Upper Paleolithic, and especially pre-ice age, populations are too sparse to comment on whether or not this is representative of a change in the genetic structure of the population, perhaps caused by a bottleneck during the LGM and a subsequent repopulation from glacial refugia.  Figure 1. Tree for 54 Present-Day Humans, Ten Ancient Modern Humans, and Seven Archaic Humans The phylogeny in the top panel was constructed using Maximum Parsimony and rooted using midpoint rooting. The branches for present-day humans do not all end at the same point, giving a sense of the inherent uncertainty in time measurements based on mtDNA due to its limited sequence span. However, the consistent shortening of the branches of ancient humans relative to their closest present-day human relatives is apparent in the figure. This is the basis for our clock calibration. Pre- and post-Neolithic ancient samples are indicated as red and blue circles, respectively, and colored squares indicate the geographical origin of 54 present-day humans that we coanalyzed with them. Date estimates for major divergence events are shown at the nodes. In the bottom panel we show a map giving geographical origin of the samples. Using ancient mtDNA sequences from securely dated archaeological samples as calibration points has allowed us to obtain an estimate of the mtDNA substitution rate that is complementary to the existing estimates based on calibration from the fossil and archaeological records. We arrive at a rate of 1.57 × 10−8 substitutions per site per year for the coding region and 2.67 × 10−8 substitutions per site per year for the whole molecule, which is approximately 1.6-fold higher than the fossil-calibrated rate [7]. Our inferred substitution rate from the whole mtDNA implies a coalescence date for all modern human mtDNAs of 120–197 kya and of 62–95 kya for hg L3, the lineage from which all non-African mtDNA hgs descend. This places a conservative upper bound of 95 kya for the time of the last major gene exchange between non-African and sub-Saharan African populations. It is important to recognize that this divergence time may merely represent the most recent gene exchanges between the ancestors of non-Africans and the most closely related sub-Saharan Africans and thus may reflect only the most recent population split in a long, drawn-out process of population separation [1]. Nevertheless, the fact that hg L3 is currently so widespread within Africa suggests that the population split dated by L3 is likely to be one of the most important ones in that history of separation, giving rise to lineages that contributed substantial fractions to the ancestry of both present-day sub-Saharan African and present-day non-African populations. Although our estimate for the population divergence of non-African and sub-Saharan Africans has a small overlap with those calculated from the de novo genomic rates, which range from 90 to 130 kya [1], the ∼30,000 year difference in the mean divergence time obtained via the different methods is notable. We believe this discrepancy is unlikely to be explained by differences in the inheritance patterns between the two parts of the genome (exclusively maternal versus biparental) or by differences in generation times of males and females [19]. In particular, de novo rates appear to scale with parental age (per year) rather than with generation time [4], and our calculation from branch shortening directly uses units of years without making any assumptions about generation times. We note that our calculated dates are more consistent with some interpretations of the fossil record: for example, the low nuclear mutation rates from the de novo investigations imply a date of human-orangutan speciation that is at least ten million years older than what is supported by the fossil record [20], whereas our dates are in accord with a more recent speciation [21, 22]. One possible reason for the discrepancies between our inferences and those that have been made based on de novo mutation rates in the nuclear genome is a substantial rate of false-negative mutations in the de novo data sets (due to the intense filtering that these studies need to apply to discriminate false-positive mutations from true positives). It is also possible that the filtering applied in the de novo mutation rate estimation studies has excluded subsets of the genome that are more mutable and that have been included in sequence divergence calculations; it is important to estimate mutation rate and sequence divergence in the same subsets of the genome to properly calibrate time estimates and this has not been done as far as we are aware. An important direction for future research is to compare the new de novo rates with those estimated based on patterns of substitutions observed over time in the nuclear genome as determined from ancient sequences [23], using methods similar to those employed here. Current Biology, Volume 23, Issue 7, 8 April 2013, Pages R286-R288 |
|

