|
|
Post by Admin on Dec 22, 2021 22:20:03 GMT
Y-STR haplotype networks and diversity We genotyped a subset of 1355 R1a samples for 10–19 Y-chromosome STR loci (Supplementary Table 3) and constructed networks for both hg R1a-Z282 and hg R1a-Z93 (Supplementary Figure 1 and Supplementary Figure 2). Although we could assign haplotypes to various haplogroups, power to identify substructure within hg R1a-M198 was limited, consistent with previous work.22, 52 Although haplotype diversity is generally very high (H>0.95) in all haplogroups (Supplementary Table 3), lower diversities occur in south Siberian paragroup R1a-Z93* (H=0.921), in Jewish R1a-M582 (H=0.844) and in Roma R1a-M780 (H=0.759), consistent with founder effects that are evident in the network patterns for these populations (Supplementary Figure 2). Origin of hg R1a To infer the geographic origin of hg R1a-M420, we identified populations harboring at least one of the two most basal haplogroups and possessing high haplogroup diversity. Among the 120 populations with sample sizes of at least 50 individuals and with at least 10% occurrence of R1a, just 6 met these criteria, and 5 of these 6 populations reside in modern-day Iran. Haplogroup diversities among the six populations ranged from 0.78 to 0.86 (Supplementary Table 4). Of the 24 R1a-M420*(xSRY10831.2) chromosomes in our data set, 18 were sampled in Iran and 3 were from eastern Turkey. Similarly, five of the six observed R1a1-SRY10831.2*(xM417/Page7) chromosomes were also from Iran, with the sixth occurring in a Kabardin individual from the Caucasus. Owing to the prevalence of basal lineages and the high levels of haplogroup diversities in the region, we find a compelling case for the Middle East, possibly near present-day Iran, as the geographic origin of hg R1a. Spatial dynamics of R1a lineage frequencies We conducted a spatial autocorrelation analysis of the two primary subgroups of R1a (Z282 and Z93) and of each of their subgroups independently (Supplementary Figure 3). Each correlogram was statistically significant. We observed clinal distributions (continually decreasing frequency with increasing geographic distance) across a large geographic area in the two macrogroups and in M558 and M780 as well. One group (Z2125) did not reveal any discernible pattern, and the analysis of four groups (Z282*, Z284, M458, and Z93*) indicated potential clinal distributions that do not extend across the full geographic range under study. Therefore, we also analyzed partial ranges for Z282* and M458 in Europe, the Caucasus, and the Middle East, and for Z284 in Europe, but these partial range analyses also failed to yield evidence of clinal distributions. We also conducted PCA of R1a subgroups (Figure 4). The first principal component explains 21% of the variation and separates European populations at one extreme from those of South Asia at the other. The second explains 14.7% of the variation and is driven almost exclusively by the high presence of M582 among some Jewish populations, particularly the Ashkenazi Jews. PC2 separates them from all other populations. When we consider haplogroups rather than populations (Supplementary Figure 4), we see that the clustering of European populations is due to their high frequencies of M558, M458, and Z282*, whereas the M780 and Z2125* lineages account for the South Asian character of the other extreme. Figure 4 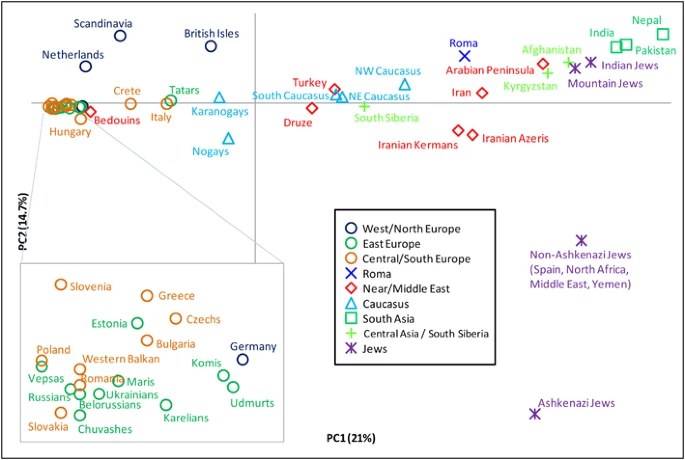 Principal component analysis of hg R1a subclades. The plot was obtained by collapsing the 126 populations into 49 regionally/culturally defined groups and calculating R1a subclade frequencies relative to R1a-M198. We excluded one population with small overall sample size and all populations in which fewer than 5 R1a Y-chromosomes were observed. To put our frequency distribution maps, PCA analyses, and autocorrelation results in archaeological context, we note that the earliest R1a lineages (genotyped at just SRY10381.2) found thus far in European ancient DNA date to 4600 years before present (YBP), a time corresponding to the Corded Ware Culture,53 whereas three DNA sample extracts from the earlier Neolithic Linear Pottery Culture (7500–6500 YBP) period were reported as G2a-P15 and F-M89(xP-M45) lineages.54 This raises the possibility of a wide and rapid spread of R1a-Z282-related lineages being associated with prevalent Copper and Early Bronze Age societies that ranged from the Rhine River in the west to the Volga River in the east55 including the Bronze Age Proto-Slavic culture that arose in Central Europe near the Vistula River.56 It may have been in this cultural context that hg R1a-Z282 diversified in Central and Eastern Europe. The corresponding diversification in the Middle East and South Asia is more obscure. However, early urbanization within the Indus Valley also occurred at this time57 and the geographic distribution of R1a-M780 (Figure 3d) may reflect this. To evaluate the potential role of R1a diversification in these post-Neolithic events, we took two approaches toward estimating the time to the most recent common ancestor (TMRCA). The first was a Y-STR-based coalescent time estimation, the results of which (Supplementary Table 5) demonstrate the unsuitability of the pedigree mutation rate, as supported also by the evidence in Wei et al,3 the ages being severely underestimated. Alternatively times based on the evolutionary mutation rate,48 which is prone to overestimation, should be regarded as the upper bounds on the sub-hg dispersals. The second approach was TMRCA estimation based on whole Y-chromosome sequencing data. |
|
|
|
Post by Admin on Dec 23, 2021 1:21:48 GMT
Whole Y-chromosome sequences from R1a and R1b: TMRCA estimates The SNPs that we genotyped across 126 populations reveal considerable information about the topology of the haplogroup tree, but they were ascertained in a biased manner, and they are too few in number to convey any meaningful branch-length information. Hence, our SNP genotyping results are devoid of temporal information. To obtain unbiased branch lengths to estimate TMRCA, we analyzed whole Y-chromosome sequences (9.99 Mb of which were usable) of 13 individuals: 8 R1a and 5 R1b. We used MEGA57 to construct a bootstrap consensus maximum likelihood tree (Figure 5) based on 928 R1 SNPs (Supplementary Data File 1), of which 462 were previously named.10 To define the ancestral and derived states of SNPs corresponding to the roots of the R1a and R1b subtrees (branches 23 and 8 in Figure 5, respectively), we called genotypes and constructed the tree jointly with previously published hg E sequences,4 which constituted an outgroup. Figure 5 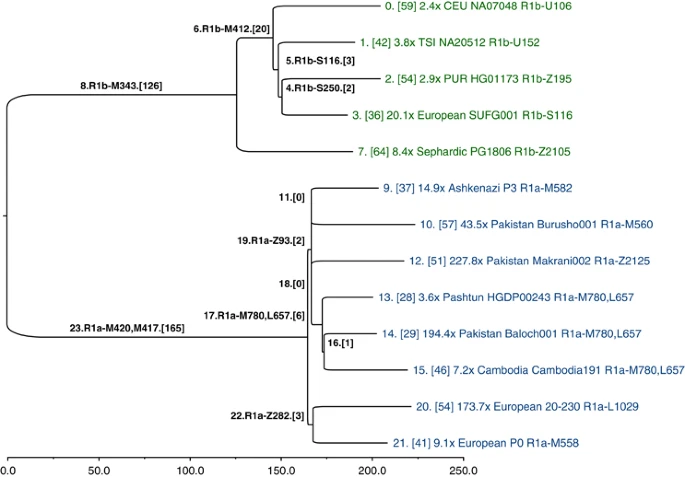 Y-chromosome phylogeny inferred from 13 ∼10-Mb sequences of hg R individuals. Branches are drawn proportional to the number of derived variants. Each of the 24 branches is labeled by an index, and the number of SNPs assigned to the branch is shown in brackets. The tips of the tree are labeled with sequencing coverage, population, ID, and the most derived commonly known SNP observed in the corresponding sample. A consensus has not yet been reached on the rate at which Y-chromosome SNPs accumulate within this 9.99 Mb sequence. Recent estimates include one SNP per: ∼100 years,58 122 years,4 151 years5 (deep sequencing reanalysis rate), and 162 years.59 Using a rate of one SNP per 122 years, and based on an average branch length of 206 SNPs from the common ancestor of the 13 sequences, we estimate the bifurcation of R1 into R1a and R1b to have occurred ∼25 100 ago (95% CI: 21 300–29 000). Using the 8 R1a lineages, with an average length of 48 SNPs accumulated since the common ancestor, we estimate the splintering of R1a-M417 to have occurred rather recently, ∼5800 years ago (95% CI: 4800–6800). The slowest mutation rate estimate would inflate these time estimates by one-third, and the fastest would deflate them by 17%. With reference to Figure 1, all fully sequenced R1a individuals share SNPs from M420 to M417. Below branch 23 in Figure 5, we see a split between Europeans, defined by Z282 (branch 22), and Asians, defined by Z93 and M746 (branch 19; Z95, which was used in the population survey, would also map to branch 19, but it falls just outside an inclusion boundary for the sequencing data4). Star-like branching near the root of the Asian subtree suggests rapid growth and dispersal. The four subhaplogroups of Z93 (branches 9-M582, 10-M560, 12-Z2125, and 17-M780, L657) constitute a multifurcation unresolved by 10 Mb of sequencing; it is likely that no further resolution of this part of the tree will be possible with current technology. Similarly, the shared European branch has just three SNPs. We caution against ascribing findings from a contemporary phylogenetic cluster of a single genetic locus to a particular pre-historic demographic event, population migration, or cultural transformation. The R1a TMRCA estimates we report have wide confidence intervals and should be viewed as preliminary; one must sequence tens of additional R1a samples to high coverage to uncover additional informative substructure and to bolster the accuracy of the branch lengths associated with the more terminal portions of the phylogeny. Although some of the SNPs on the lineages we have defined by single SNPs are undoubtedly rare (eg, the Z2125 sub-hg M434, Figure 1, Supplementary Table 4), it remains possible that future genotyping effort using the SNPs in Supplementary Data File 1 may expose other substructure at substantial frequency, commensurate with more recent episodes of population growth and movement. In addition, high coverage sequences using multiple male pedigrees sampled across various haplogroups in the global Y phylogeny will be needed to more accurately estimate the Y-chromosome mutation rate. Nonetheless, despite the limitations of our small sample of R1a sequences, the relative shortness of the branches and their geographic distributions are consistent with a model of recent R1a diversification coincident with range expansions and population growth across Eurasia. Conclusion Our phylogeographic data lead us to conclude that the initial episodes of R1a-M420 diversification occurred in the vicinity of Iran and Eastern Turkey, and we estimate that diversification downstream of M417/Page7 occurred ∼5800 years ago. This suggests the possibility that R1a lineages accompanied demic expansions initiated during the Copper, Bronze, and Iron ages, partially replacing previous Y-chromosome strata, an interpretation consistent with albeit limited ancient DNA evidence.54, 60 However, our data do not enable us to directly ascribe the patterns of R1a geographic spread to specific prehistoric cultures or more recent demographic events. High-throughput sequencing studies of more R1a lineages will lead to further insight into the structure of the underlying tree, and ancient DNA specimens will help adjudicate the molecular clock calibration. Together these advancements will yield more refined inferences about pre-historic dispersals of peoples, their material cultures, and languages.57, 61, 62 |
|
|
|
Post by Admin on Dec 23, 2021 19:00:57 GMT
Separating the post-Glacial coancestry of European and Asian Y chromosomes within haplogroup R1a
Abstract
Human Y-chromosome haplogroup structure is largely circumscribed by continental boundaries. One notable exception to this general pattern is the young haplogroup R1a that exhibits post-Glacial coalescent times and relates the paternal ancestry of more than 10% of men in a wide geographic area extending from South Asia to Central East Europe and South Siberia. Its origin and dispersal patterns are poorly understood as no marker has yet been described that would distinguish European R1a chromosomes from Asian. Here we present frequency and haplotype diversity estimates for more than 2000 R1a chromosomes assessed for several newly discovered SNP markers that introduce the onset of informative R1a subdivisions by geography. Marker M434 has a low frequency and a late origin in West Asia bearing witness to recent gene flow over the Arabian Sea. Conversely, marker M458 has a significant frequency in Europe, exceeding 30% in its core area in Eastern Europe and comprising up to 70% of all M17 chromosomes present there. The diversity and frequency profiles of M458 suggest its origin during the early Holocene and a subsequent expansion likely related to a number of prehistoric cultural developments in the region. Its primary frequency and diversity distribution correlates well with some of the major Central and East European river basins where settled farming was established before its spread further eastward. Importantly, the virtual absence of M458 chromosomes outside Europe speaks against substantial patrilineal gene flow from East Europe to Asia, including to India, at least since the mid-Holocene.
Introduction
Human populations across the world are characterized by generally low genetic differences as compared with their intrapopulation variation. These differences can be quantitative, pronounced in different frequencies of the same derived states of ancient polymorphic markers (eg, majority of the HapMap markers1), or qualitative, in which case younger derived variants are found restricted to a particular geographic region or population. The Y-chromosome haplogroup structure frequently shows a good qualitative correlation with continental boundaries, and the geographic specificity of the markers can most often be explained by their phylogenetic descent order rather than by drift alone.2, 3 Recently evolved polymorphisms unless amplified by selection or specific founder effects tend to have low frequencies in modern populations, characterized generally by increased effective population sizes in the Holocene period. One of the notable outliers to this rule, because of its high frequency and young age, is the transcontinental spread of haplogroup R1a.4, 5 Early observations have led to various interpretations associating R1a phylogeography with certain cultural developments of the past. Even though R1a occurs as the most frequent Y-chromosome haplogroup among populations representing a wide variety of language groups, such as Slavic, Indo-Iranian, Dravidian, Turkic and Finno-Ugric, many authors have been particularly interested in the link between R1a and the Indo-European language family. For example, R1a frequency patterns have been discussed6, 7 in the context of the purported link connecting Indo-European-speaking pastoralists and the archeological evidence on the distribution of the Kurgan culture in the Pontic steppe.8 A more precise interpretation of the underlying prehistoric and historic episodes of R1a chromosomes across this wide span of Eurasian geography remains largely unknown because of insufficient information on the phylogenetic subdivisions within haplogroup R1a. We address this shortcoming here by analyzing more than 11 000 DNA samples from across Eurasia, including more than 2000 from haplogroup R1a to ascertain the phylogenetic information of the newly discovered R1a-related SNPs. We also examine the STR diversity of the associated R1a subclades to better understand the demographic history and prehistoric cultural associations of one of the most widely spread and frequent Y-chromosome haplogroups in the world with post-Last Glacial Maximum origin.
Materials and methods
Twelve recently reported R1a markers ascertained in one R1a1 individual2, 9 across extensive but unspecified coverage and two new SNPs discovered in two R1a1 individuals during a scan of ∼44 kb10 were genotyped by denaturing high-performance liquid chromatography (DHPLC) and confirmed by direct sequencing in an initial screening of 18 DNA samples belonging to haplogroup R1a from different geographic regions spanning Scandinavia to India. Twelve of these markers were derived in all individuals carrying the M17 mutation, whereas one of the markers, Page68, exhibited an ancestral allele in all samples and was therefore not evaluated further. In addition, two new SNPs were discovered. One (M434) while surveying another SNP reported in the flanking sequence of DYS43811 by DHPLC in a globally representative collection of DNAs that included individuals from Pakistan, and another (M458) was discovered during the initial survey of the Hinds et al9 rs17250901 homopolymer variant. Markers M434 and M458 were variable in a subset of the 18 R1a screening samples and represent new informative subclades of R1a1. Another SNP (M334) was ascertained previously by DHPLC in one Estonian in a panel of 48 R1a1 samples. Marker M334 was not observed in an additional survey of 100 R1a1 Estonian samples and was not studied further. In the population surveys, the markers were genotyped either by DHPLC, RFLP or TaqMan (Applied Biosystems, Foster City, CA, USA) assays. Within specific haplogroups, median-joining networks were constructed. Specifications for the analyses are detailed in the relevant figure legends. The age of microsatellite variation within haplogroups was evaluated using the methodology described by Zhivotovsky et al12 as modified according to Sengupta et al13 using microsatellite evolutionary effective mutation rate of 6.9 × 10−4 per 25 years. Sample sizes and frequencies of the main R1a subclades are reported in Supplementary Tables 1–3. STR haplotype data are given in Supplementary Tables 4, 6 and 7. Supplementary Table 5 reports the primer sequences used in genotyping the informative SNPs.
|
|
|
|
Post by Admin on Dec 23, 2021 20:04:05 GMT
Results and discussion By using the new SNP markers, we were able to fractionate the R1a defining node into a nested series of branches that are reinforced by multiple phylogenetically equivalent mutations (inset, Figure 1). All chromosomes unresolved previously beyond the R1-M173* level14, 15, 35, 36 that were available to us are now attributed to either R1a*-M420 or R1b*-M343 haplogroups. Consequently, we revise the haplogroup nomenclature following the YCC guidelines.3, 5 Although the occurrences of the most basal haplogroup R1a*-M420(xSRY10831.2) and the intermediate haplogroup R1a1*-SRY10831.2(xM17) are rare (Supplementary Table S1), the descendent haplogroup R1a1a-M17 assemblage displays informative frequencies above a few percent in populations comprising a broad expanse of Eurasian geography ranging from Norway and Northeast Asia to south India, whereas frequencies above 10% occur in East Europe, West, South and central Asia (Supplementary Table S2, Figure 1). With the exception of a few localized low-frequency subhaplogroups,4, 14, 37 the majority of haplogroup R1a1a chromosomes have remained so far phylogenetically indistinct.  Figure 1 Geographic distribution of haplogroup R1a1a frequency. Spatial frequency map was obtained applying the frequencies from Supplementary Table S2 and for 8429 individuals representing 118 populations from literature.7, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 Dots on the map indicate the approximate locations of the sampled populations. The frequency data were converted to isofrequency maps in Surfer software (version 7, Golden Software Inc., Golden, CO, USA) following the Kriging procedure. The inset map illustrates the available data (Supplementary Table S2) for the regional expansion times in KYA (thousands of years ago) of M17 Y-chromosomes. We note that especially in the latter case the density of the data points is too low for any viable geostatistical analyses. Phylogenetic tree relating SNP markers that define haplogroup R1a and its subgroups is shown in the inset. Previously described SNP markers3 are underlined. Markers M56, M157, M64.2, M87, M204, P98 and PK5 shown in gray font were not typed as they were previously detected at nonpolymorphic frequencies in other studies. PCR amplicons for 12 SNPs from Hinds et al9 (M420, M448, M449, M459, M511, M513, M516 and rs17250901) and 2 from Repping et al10 (Page07 and Page68) were designed and tested for male specificity using female control DNA. The phylogenetic relationships of these SNPs were evaluated in a geographically diverse panel of 18 R1a1 samples and 2 R1b* samples ranging from Northwest Europe to South Asia using DHPLC technology, and confirmed by direct sequencing of representative samples. Detailed specifications for these markers are given in Supplementary Table S5. Recent Arabian Sea gene flow The marker M434, defining the novel Y-chromosome haplogroup R1a1a6, was observed altogether in 14 individual samples in our screening of 691 R1a1a chromosomes (Supplementary Table S3). Given these data, the haplogroup R1a1a6 distribution seems to be restricted mainly to Pakistan whereas the Omani R1a1a6 samples, all three of which share the same STR haplotype, indicate recent gene flow across the Persian Gulf. The low STR haplotype diversity of R1a1a6 and its absence in 212 Indian R1a1a samples suggest that the M434 mutation may have arisen recently in Pakistan. In situ diversification in Central Europe In contrast to the restricted geographic pattern of M434, the R1a1a7 defining marker, M458, was found to be variable in a number of populations, and thus it provides the first significant geographic compartmentalization within the overarching haplogroup R1a distribution. The haplogroup R1a1a7 distribution is confined to Central and Eastern Europe and does not extend eastward beyond the Ural Mountains or southward beyond Turkey (Supplementary Table S2, Figure 2). Its spread in the Caucasus is specific: although absent in the Dagestanian group, it is present at low frequencies both in the northwestern and southern populations, and in particular in Karanogays, who only relatively recently were spread as pastoral nomadic people alongside the Ponto-Caspian steppe belt. The highest frequency of haplogroup R1a1a7 (over 30%) is observed in Central and Southern Poland. Frequencies higher than 10% occur among Western and Eastern Slavic populations whereas elsewhere in Europe, including Southern Slavic groups, the frequency of the derived M458G allele decreases rapidly away from its frequency peak that coincides broadly with the overall R1a1a frequency maximum in Poland (Figures 1 and and2).2). The R1a1a*(xM458) chromosomes on the other hand are less frequent in Poland and display frequency maximums in Belarus and southwest Russia (Supplementary Table S2).  Figure 2 Geographic distribution of haplogroup R1a1a7-M458 frequency. The spatial frequency map was obtained applying the frequencies from Supplementary Table S2 (dots on the map indicate the approximate locations of the sampled populations) to the Surfer software (version 7, Golden Software Inc., Golden, CO, USA) following the Inverse Distance to Power (Power 3.75; smoothness 0) procedure with added break lines indicated by dashed blue lines in the seas. Spatial distribution of the expansion times of the regional M458 derived Y-chromosomes is shown in the lower left inset map according to data in Supplementary Table S4. See text for discussion concerning the spread of M458 lineages with the major European river basins (shown in blue) and major Neolithic and Metal Age cultures. Analysis of associated STR diversity profiles revealed that among the R1a1a*(xM458) chromosomes the highest diversity is observed among populations of the Indus Valley yielding coalescent times above 14 KYA (thousands of years ago), whereas the R1a1a* diversity declines toward Europe where its maximum diversity and coalescent times of 11.2 KYA are observed in Poland, Slovakia and Crete. As islands such as Crete have been subject to multiple episodes of colonization from different source regions, it is not inconsistent that R1a1a* Td predates the date of its first colonization by the first farmers approximately 9 KYA.38 Also noteworthy is the drop in R1a1a* diversity away from the Indus Valley toward central Asia (Kyrgyzstan 5.6 KYA) and the Altai region (8.1 KYA) that marks the eastern boundary of significant R1a1a* spread (Figure 1, Supplementary Table S4.). In Europe, Poland also has the highest R1a1a7-M458 diversity, corresponding to approximately an 11 KYA coalescent time (Supplementary Table S4). Other populations in Europe exhibit declining diversity when sampled at increasing distance away from Central Europe (Figure 2). Westward of the Rhine overall R1a1a frequency is low, signaling a genetic boundary with R1b varieties.39 However, the patterns of currently observed Y-chromosome diversity in East/Central Europe are unlikely to be explained solely by population movements of the last century.40 Although the median STR haplotype of the derived M458G allele differs from the median type of the ancestral M458A chromosomes at 3 of the 10 STR loci considered in our analyses, the STR data alone are not informative for unambiguous inference of whether an individual has the A or G allele (Supplementary Figures S1 and S2) underscoring the extent of STR saturation and the importance of SNP genotyping to assess phylogenetic ancestry even among closely related lineages. |
|
|
|
Post by Admin on Dec 23, 2021 21:15:01 GMT
Phylogeography
Haplogroup frequency, haplotype diversity and coalescent times are three parameters that can be considered as informative for making inferences about the origins and polarity of spread of alleles among populations. The most distantly related R1a chromosomes, that is, both R1a* and R1a1* (inset, Figure 1), have been detected at low frequency in Europe, Turkey, United Arab Emirates, Caucasus and Iran14, 41 (Supplementary Table S1). The highest STR diversity of R1a1a*(xM458) chromosomes are observed outside Europe, in particular in South Asia (Figure 1, Supplementary Table S4), but given the lack of informative SNP markers the ultimate source area of haplogroup R1a dispersals remains yet to be refined.
In Europe a large proportion of the R1a1a variation is represented by its presently identified subclade R1a1a7-M458 that is virtually absent in Asia. Its major frequency and relatively low diversity in Europe can be explained thus by a founder effect that according to our coalescent time estimation falls into the early Holocene period, 7.9±2.6 KYA (Supplementary Table S4). The highest regional date of 10.7±4.1 KYA among Polish R1a1a7 carriers falls into the period of recolonization of this region by Mesolithic (Swiderian and subsequent cultures) settlers.42, 43 The time window of 10−5 KYA BP is a culturally complex juncture period between the Mesolithic and early Neolithic in Europe, thus, not allowing us to relate founder effect with any particular culture specifically. Most broadly, the autochthonous European origin of haplogroup R1a1a7, its narrow spatial distribution and the inversely related decreasing expansion times with increased distance from its core frequency and diversity area are suggestive of a notably successful demic expansion starting from a small subset of radiating founder lineages during the early Holocene period. It should be noted, though, that the inevitably large error margins of our coalescent time estimates do not allow us to exclude its association with the establishment of the mainstream Neolithic cultures, including the Linearbandkeramik (LBK), that flourished ca. 7.5−6.5 KYA BP in the Middle Danube (Hungary) and was spread further along the Rhine, Elbe, Oder, Vistula river valleys and beyond the Carpathian Basin.44
Migratory and early agricultural zones
River valleys are migratory corridors for organisms including humans and such riparian habitats provide opportunities for the forager lifestyle, settled agriculture and establishment of trade networks. The Neolithic communities in Central Europe were primarily located on the margins of river valleys with fertile soils at elevations less than 500 m.45 Haplogroup R1a1a7-M458 diversity and frequency are highest in river basins known to be associated with several early and late Neolithic cultures (Figure 2, Supplementary Figure S3). Assuming the founder effect we detect originated in the sparse Mesolithic population of Central-North Europe, the genetic evidence suggests strong cultural interaction and admixture occurred between the pioneer horticultural groups and local foragers, which resulted in widespread adaptation of the Neolithic lifestyle by indigenous residents. This interpretation is consistent with computational models indicating that although the process of the expansion of farming communities throughout much of Europe would have been demic, even minute amounts of gene flow from foragers over a long time period would have lead to a predominantly Mesolithic contribution to their admixed offspring.46 Following this model, it would not be surprising to associate a localized Neolithic demic expansion with a genetic lineage absent in the Fertile Crescent where farming originated and where other Y-chromosome haplogroups, such as G and J, have been associated with the initial demic spread of farming toward Southeast Europe.38 However, it should be noted that ancient mtDNA evidence from the Central European Mesolithic and LBK sites shows a lack of substantial continuity between Mesolithic, Neolithic and presently living populations of the area.47, 48 Notably, mtDNA haplogroups R1a, U4, U5, HV3 and HV4, which have been inferred to have pre-Neolithic spread in East Europe, occur at marginally low frequencies in India.49
It is noteworthy that the LCT-13910T allele associated with lactase persistence and agricultural pastoralism overlaps broadly with the spatial distribution50 of the derived M458G allele. Direct ancient DNA evidence suggests that the lactase persistence allele would have reached high frequency in this area, likely due to strong positive selection, only after the LBK period.51 However, computer simulations have shown that its increased frequency particularly in North Europe does not necessarily imply stronger effect of positive selection there than in other parts of Europe.52 Ancient DNA evidence for the Y-chromosome M458G allele is still lacking and it is therefore possible only to speculate about its existence and prevalence in Neolithic Europe. Beyond its spread in the Central European river basins (Figure 2), the LBK extended around the northern Carpathians into the steppe zone of Ukraine and participated in the establishment of the Criş culture.53 Our data showing high frequency of R1a1a north of the Carpathians and its lower frequency to the South, in the Tisza river valley, are consistent with the genetic boundary previously reported for this region.16
Copper and Bronze age parallels
Figure 2 also shows a remarkable geographic concordance of the R1a1a7-M458 distribution with the Chalcolithic and Early Bronze Age Corded Ware (CW) cultures of Europe that prospered from ca. 5.5−4.5 KYA BP.54 Ancient DNA evidence from a 4600-year-old multiple burial unearthed near Eulau, Germany and attributed to the Central European CW culture, identified the remains of three males carrying the SRY10831.2 mutation and sharing the same YSTR haplotype, implying a single family lineage.55 Although haplogroup affiliation cannot be inferred with certainty from STR data alone, a composite 15-locus YSTR haplotype representing the ancient lineage suggests its potential R1a1a*(xM458) membership due to four alleles (DYS391=11, DYS439=10, DYS389B=17 and DYS458=15) shared with the median R1a1a*(xM458) haplotype (Supplementary Tables S4 and S7). Interestingly, from the list of regional median haplotypes, the ancient haplotype is most similar to the German R1a1a*(xM458) type.
Indo-Europeans
A final comment can be made concerning the relationship between R1a phylogeography and contested origin of Indo-Europeans that is generally, though not solely, attributed to either Anatolia, the South Caucasus or the North Pontic-Caspian regions (Gray and Atkinson56 and references therein). Haplogroup R1a1a occurs in all three of these areas and beyond at informative frequencies (Figure 1). Consistent with its wide geographic spread, the coalescent time estimates of R1a1a correlate with the timing of the recession of the Last Glacial Maximum and predate the upper bound of the age estimate of the Indo-European language tree. Although virtually absent among Romance, Celtic and Semitic speakers, the presence and overall frequency of haplogroup R1a does not distinguish Indo-Iranian, Finno-Ugric, Dravidian or Turkic speakers from each other. Some contrast, however, is unfolding in its subclade frequencies. Although the R1a1a* frequency and diversity is highest among Indo-Aryan and Dravidian speakers, the subhaplogroup R1a1a7-M458 frequency peaks among Slavic and Finno-Ugric peoples. Although this distinction by geography is not directly informative about the internal divisions of these separate language families, it might bear some significance for assessing dispersal models that have been proposed to explain the spread of Indo-Aryan languages in South Asia as it would exclude any significant patrilineal gene flow from East Europe to Asia, at least since the mid-Holocene period.
|
|

