|
|
Post by Admin on Jan 14, 2019 18:46:28 GMT
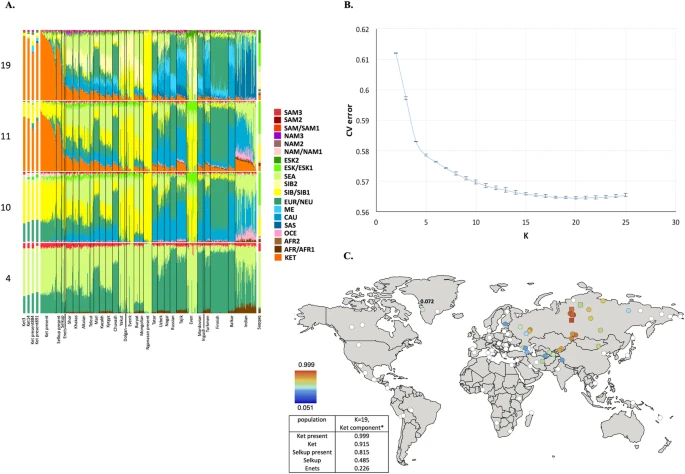 Figure 1 (A) Admixture coefficients plotted for dataset ‘GenoChip + Illumina arrays’. Abbreviated names of admixture components are shown on the left as follows: SAM, South American; NAM, North American; ESK, Eskimo (Beringian); SEA, South-East Asian; SIB, Siberian; NEU, North European; ME, Middle Eastern; CAU, Caucasian; SAS, South Asian; OCE, Oceanian; AFR, African. The Ket-Uralic (‘Ket’) admixture component appears at K ≥ 11, and admixture coefficients are plotted for K = 4, 10, 11, and 19. Although K = 20 demonstrates the lowest average cross-validation error, the Ket-Uralic component splits in two at this K value, therefore K = 19 was chosen for the final analysis. Only populations containing at least one individual with >5% of the Ket-Uralic component at K = 19 are plotted, and individuals are sorted according to values of the Ket-Uralic component. Admixture coefficients for the Saqqaq ancient genome are shown separately on the right, and for two reference Kets and two Ket individuals from this study - on the left. (B) Average cross-validation (CV) error graph with standard deviations plotted. Ten-fold cross-validation was performed. The graph has a minimum at K = 20. (C) Color-coded values of the Ket-Uralic admixture component at K = 19 plotted on the world map using QGIS v.2.8. Maximum values in each population are taken, and only values >5% are plotted. Top five values of the component are shown in the bottom left corner, and the value for Saqqaq is shown on the map. Ket-Uralic’ admixture component Using the GenoChip SNP array20, we genotyped 130 K ancestry-informative markers in the Ket, Selkup, Nganasan, and Enets populations (Suppl. file S1). Following the exclusion of first-, second-, and third-degree relatives among the individuals genotyped in this study (Suppl. file S1, Suppl. Fig. 4.1), we merged the GenoChip array data with the published SNP array datasets to produce a worldwide dataset of 90 populations and 1,624 individuals, focused on Siberia and America (Suppl. Table 2). The intersection dataset, containing 32,189 SNPs (Suppl. Table 1), was analyzed with ADMIXTURE37 (Fig. 1). At K ≥ 11, ADMIXTURE identified a characteristic component for the Ket population (Suppl. Information Section 5). This component reached its global maximum of nearly 100% in Kets, closely followed by Selkups from this study (up to 81.5% at K = 19), the reference Selkups (up to 48.5%) and the Enets (up to 22.6%). The difference between the Selkups from this study and the reference Selkups21 can be attributed to a much closer geographic proximity of the former population to the settlements of Kets, with whom they have a long history of cohabitation and mixture2,10.  The ‘Ket’ component occurred at high levels (up to ~20%) in four Turkic-speaking populations of the Altai region: Shors, Khakases, Altaians, and Teleuts. Notably, the Altai region was populated by Yeniseian-speaking people before they were forced to retreat north (Suppl. Information, Section 2). Lower levels of the ‘Ket’ component, from 5% to 15%, were observed in the following geographic regions (in decreasing order): the Volga-Ural region, Central and South Asia, East Siberia and Mongolia, and North Caucasus. The ‘Ket’ component also occurred at a low level in Russians (up to 7.1%), Finns (up to 5.4%), and, remarkably, in the Saqqaq ancient genome from Greenland (7.2%, see below). In order to verify and explain the geographic distribution of the ‘Ket’ admixture component, we have performed ADMIXTURE analysis on two additional datasets, different in population (Suppl. Table 2) and marker selection (Suppl. Table 1) (see Suppl. Information, Section 5). In summary, we suggest the existence of an admixture component with a peculiar geographic distribution, observed in some previous studies but not discussed there17,18. In addition to the Kets, this component is characteristic also for Samoyedic-speaking and Ugric-speaking people of the Uralic language family: Selkups, Enets, Nenets, Khanty, Mansi, with a notable exception of Samoyedic-speaking Nganasans. The proportion of the ‘Ket-Uralic’ admixture component correlated strongly with the worldwide frequency of mitochondrial haplogroup U4 (Pearson’s correlation coefficient up to 0.8 and a corresponding p-value of 7 × 10−8) and with the frequency of Y-chromosomal haplogroup Q in Eurasian populations (correlation coefficient up to 0.9 and p-value 2 × 10−7) (Suppl. Information, Section 10). 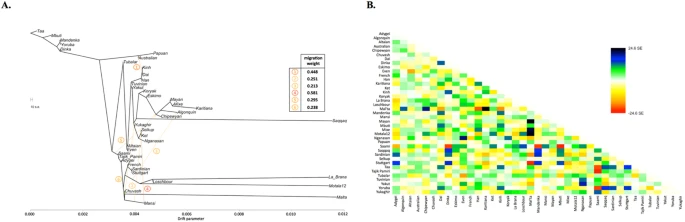 Figure 2 Kets in the context of Siberian populations In order to study the relationship of Kets and other Siberian populations with the relevant ancient genomes, we have constructed three additional datasets: the dataset based on the Ket genome sequences and the HumanOrigins array SNP data22, and two datasets based on genome sequences only (Suppl. Tables 1 and 2). The Ket and Selkup populations were closely related according to multiple analyses (see the ADMIXTURE plot in Fig. 1, PCA plots in Suppl. Figs. 6.3, 6.6, TreeMix tree in Fig. 2, and outgroup f3 statistics38 in Suppl. Fig. 7.2). Nganasans appeared as the closest relatives of Kets according to statistics f3 (Yoruba; Ket, X): the statistic for Nganasans was significantly different from that of the second-best hit (Suppl. Fig. 7.2). In general, outgroup f3 statistics (Yoruba; Test, X) were tightly correlated between the Kets, the Selkups, and the Nganasans, with Pearson’s correlation coefficients ranging from 0.96 to 0.999 (Suppl. Information, Section 7), suggesting that these populations form a closely related group. In line with these results, Nganasans, Kets, Selkups, and Yukaghirs formed a clade in a maximum likelihood tree constructed with TreeMix on a HumanOrigins-based dataset of 194,750 SNPs (Fig. 2). In our ADMIXTURE analyses (Fig. 1A, Suppl. Fig. 5.4), the Saqqaq Paleo-Eskimo individual featured the following components: Beringian, Siberian, and South-East Asian. Thus, Saqqaq Paleo-Eskimo has mostly Beringian ancestry (similar to modern Eskimo, Inuits, Aleutians, Koryaks, etc.): see outgroup f3 statistics and associated Zdiff scores in Suppl. Figs. 7.17–7.19, migration edges modelled with TreeMix in Fig. 3, and the ADMIXTURE results in the original study16. Beringian ancestry in Saqqaq is combined with considerable Siberian ancestry: 32% or 28% as a sum of Siberian ADMIXTURE components in this study (Fig. 1A, Suppl. Fig. 5.4); ~25% according to ADMIXTURE analysis in the original study (Rasmussen et al. 2010)16; from 31% to 57% according to f4 statistic ratios calculated with various outgroups (Suppl. Information, Section 8, Suppl. Table 6). This ‘core Siberian’ component in Saqqaq is apparently most closely related to modern Nganasans16 (Suppl. Fig. 7.17) and to the Nganasan-related clade in general (see a TreeMix tree in Fig. 2). The Kets are the only representatives of this clade in the genome-based datasets in this study. According to the pairwise correlation between outgroup f3 statistics (the method used in Allentoft et al.6), Kets are closer to Saqqaq as compared to Nivkhs, Altaians, Buryats, and Yakuts (Suppl. file S2). According to Euclidean distances in the ten-dimensional space of principal components on the HumanOrigins dataset, Kets were a closer population to Saqqaq than Nganasans, Selkups, Yukaghirs, and the other populations (Fig. 4). However, the outgroup f3 statistics (Yoruba; Saqqaq, Ket) in many cases were not significantly different from f3 (Yoruba; Saqqaq, other Siberian population): e.g., see |Zdiff| scores < 3 for f3 (Yoruba; Saqqaq, Nivkh) (Suppl. Figs. 7.18, 7.19). The same result was produced with f4 (Saqqaq, Yoruba; Nivkh, Ket): an absolute Z-score was lower than 2 (Suppl. Fig. 8.18A,B). |
|
|
|
Post by Admin on Jan 14, 2019 22:13:54 GMT
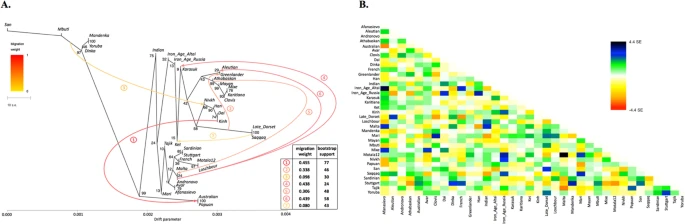 Figure 3 Allentoft et al.6 have shown that in the early Bronze Age the Altai region was inhabited by a genetically West Eurasian population of the Afanasievo archaeological culture, most similar to the Yamnaya culture with about 50% of ANE ancestry6, and to the modern Avars (Suppl. file S2, Fig. 3, Suppl. Figs. 9.4, 9.5). In the late Bronze Age this population apparently had being gradually admixed with Siberians, giving rise to the Karasuk culture and the later cultures of the Iron Age6. The ancient genome ‘Iron Age Russia’, carbon-dated to 721–889 AD6, is most closely related to the typical modern Siberians: Nganasans according to the outgroup f3 statistic in the original study6 and Altaians or Koryaks according to the outgroup f3 statistics and their pairwise correlations on our datasets lacking Nganasans (Suppl. file S2). However, according to various analyses, the ‘Iron Age Altai’ (dated roughly to 2900-1500 YBP) and the Karasuk (carbon-dated to 3531-3261 YBP) populations of two and six genomes, respectively, are most closely related to each other and to Kets (Suppl. files S2, S3, Suppl. Figs. 7.5, 7.6, 8.17A, 9.1). The outgroup f3 statistics (Yoruba; Karasuk, X) on both genome-based datasets selected Kets as the best hit for Karasuk (Suppl. files S2, S3, Suppl. Fig. 7.5), although statistics for Mayans, Greenlanders, Mixe, Saqqaq, Mal’ta, Iron Age Russia, and Aleutian were not significantly different (|Zdiff| score < 3). Similarly, Native American, Beringian populations and Selkups were the best hits for Iron Age Altai and Karasuk according to the outgroup f3 statistics in the original study (Kets were lacking in the dataset, see Allentoft, et al.6). Importantly, the Karasuk culture has been tentatively associated with the Yeniseian-speaking people based on the toponymic evidence5, and the Altai region is considered to be the homeland of the Yeniseian language family2. As another piece to this puzzle, we observed genetic continuity between the Kets and the ancient genomes from the Altai. 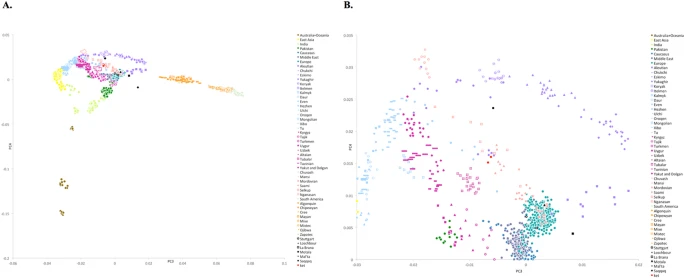 Figure 4 Mal’ta (ancient North Eurasian) ancestry in Kets The outgroup statistic f3 (Yoruba; Mal’ta, Ket) (Raghavan et al. 2014)21 was higher than statistics for all other Siberian and most Beringian populations. However, the statistic values were not significantly different within a large group of North Eurasians, according to Zdiff scores (Suppl. Figs. 7.13–7.16). Zdiff score for f3 (Yoruba; Mal’ta, Ket) vs. f3 (Yoruba; Mal’ta, Nganasan) equaled 7.4, 7.0 vs. f3 (Yoruba; Mal’ta, Yukaghir), and only 2.7 vs. f3 (Yoruba; Mal’ta, Selkup) (Suppl. Fig. 7.13). Thus, we suggest that, unlike the other members of the Nganasan-related clade (Fig. 2), Kets and, to a lesser extent, Selkups have a high proportion of Mal’ta ancestry, alternatively referred to as the ANE ancestry22. The Mal’ta ancestry in Kets was further supported by the TreeMix39 analysis, specifically by a migration edge connecting Mal’ta to the Ket-Karasuk clade, with a weight of 43% (Suppl. Fig. 9.1, Suppl. Table 7). Taking into account the admixture coefficients for the two sequenced Ket individuals (Ket891 and Ket884, Fig. 1A), we selected Ket891 as an individual with lower values of the North European and Siberian admixture components (in the K = 19 dimensional space). In addition, Ket891 was identified as non-admixed by reAdmix analyses (Suppl. Table 3). Ket891 demonstrated a slightly closer genetic affinity to Mal’ta (compare Suppl. Figs. 7.13 and 7.14). These results were consistent with calculations of f4 statistic in two configurations: (X, Chimp; Mal’ta, Stuttgart) or (X, Papuan; Sardinian, Mal’ta), reproducing the previously used statistics22,26 (Suppl. Figs. 8.1–8.6). f4 (X, Chimp; Mal’ta, Stuttgart) analysis tests whether the population X has more drift shared with Mal’ta or with Stuttgart (an early European farmer, EEF22). Sardinians were used as the closest modern proxy for EEF22 in f4 (X, Papuan; Sardinian, Mal’ta). All possible population pairs (X,Y) were tested by f4 (Mal’ta, Yoruba; Y, X) on the genome-based dataset, including both Ket individuals (Fig. 5A). Compared to Kets, Mal’ta was significantly closer to none of the populations including Native Americans. 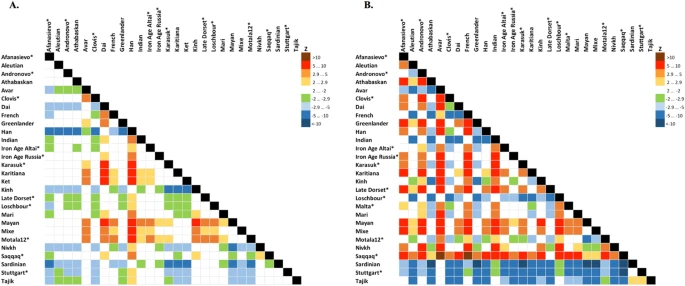 Figure 5 Based on all analyses, we can tentatively model Kets as a two-way mixture of broadly defined East Asians and ANE. Therefore, ANE ancestry in Kets can be estimated, using various f4-ratios, at 27% to 43% (depending on reference populations and datasets), vs. 25–53% in various Native American groups (Suppl. Table 6, see details in Suppl. Information, Section 8). Given non-significant Zdiff scores and f4 statistics (Fig. 5A) discussed above, it is difficult to identify the exact Eurasian population west of Chukotka and Kamchatka with the highest degree of the Mal’ta ancestry, but Kets are a good candidate. We speculate that ANE component was acquired by ancestors of Kets in the Altai region, where the Bronze Age Okunevo culture was located, with a surprisingly close genetic proximity to Mal’ta6. Later, the Yeniseian-speaking people occupied this region until the 16th–18th centuries3,4. Based on previous studies16,25,28, the Saqqaq individual, and the Paleo-Eskimos in general25 may represent a separate and relatively recent migration into America. The Paleo-Eskimos have large proportions of Beringian (i.e. Chukotko-Kamchatkan and Eskimo-Aleut), Siberian, and South-East Asian ancestry. We have also shown that Kets and Selkups belong to a group of modern populations closest to an ancient source of Siberian ancestry in Saqqaq. This group also includes, but is probably not restricted to, Uralic-speaking Nganasans and Yukaghirs (the latter speak an isolated language). Unlike the other populations of this group, Kets, and, to a lesser degree Selkups, have a high proportion of Mal’ta (ancient North Eurasian) ancestry.  As shown previously28, Chipewyans, a modern Na-Dene-speaking population, have about 16% of Saqqaq ancestry. Thus, a gene flow dated at 5,000-6,000 YBP16 can be traced from the cluster of Siberian populations to Saqqaq, and from Saqqaq to Na-Dene. However, the genetic signal in contemporary Na-Dene-speaking ethnic groups is substantially diluted. The genetic proximity of Kets to the source of Siberian ancestry in Saqqaq correlates with the hypothesis that Na-Dene languages of North America are specifically related to Yeniseian languages of Siberia, now represented by Ket language only10. However, this genetic link is indirect and requires further study of population movement and language shifts in Siberia. In addition, we show that Kets represent a modern Siberian population closest to ancient individuals of the Karasuk culture, spanning a period from about 3400 YBP to 2900 YBP (the individuals analyzed were dated to 3531-3261 YBP), and to few investigated Iron Age individuals of the Altai region (2900-1100 YBP)6. This genetic continuity correlates with historical linguistic data suggesting that the homeland of Yeniseian languages was located in the Altai region2,5. Scientific Reports volume 6, Article number: 20768 (2016) |
|
|
|
Post by Admin on Mar 28, 2019 18:02:39 GMT
 Siberia is a vast geographical region of Russia located to the east of Ural Mountains. Understanding population history of people traditionally occupying Siberia and the Trans-Uralic region (the territory to the west and east of the Ural Mountains) is of great historical interest and would shed light on origins of both modern-day Eurasians and populations of the New World. Recent studies demonstrated the potential of whole-genome sequencing for detecting genetic relationships between various European and Asian populations and identifying genetic links between modern and ancient inhabitants from these regions (Der Sarkissian et al. 2013; Lazaridis et al. 2014; Raghavan et al. 2014). However, large-scale population genetic mapping efforts such as the 1000 Genomes Project (The 1000 Genomes Project Consortium 2012) and HapMap (The International HapMap 3 Consortium 2010) have not surveyed genetic landscapes of populations from Russia. Siberia has been inhabited by hominins for hundreds of thousands of years, with some of the known archeological sites being older than 260,000 yr (Waters et al. 1997). Neanderthals inhabited Europe and Siberia until ∼40,000 yr ago (40 kya) (Hublin 2009), and anatomically modern humans expanded to these regions 60–40 kya (Hublin 2012). Traces of habitation of anatomically modern humans in Siberia date to at least 45 kya, based on a bone recently discovered near an Ust’-Ishim settlement in Western Siberia (Fu et al. 2014). The Ust’-Ishim's genome provided evidence for ancient human and Neanderthal admixture that occurred ∼50–60 kya (Fu et al. 2014). Other ancient human sites in Siberia yielded ancient DNA from ancient North Eurasians (ANEs) (Lazaridis et al. 2014), including the Upper Paleolithic 24,000-yr-old Siberian Mal'ta boy MA-1 (Raghavan et al. 2014) and the 17,000-yr-old Siberian AG-2 (Raghavan et al. 2014). Analysis of these genomes revealed genetic contribution from the ANE people to the genetic makeup of Western Siberians, Europeans, and early indigenous Americans (Lazaridis et al. 2014; Raghavan et al. 2014).  Substantial changes in the Eurasian genetic landscape took place during the Bronze Age (around 5–3 kya), a period of major cultural changes involving large-scale population migrations and replacements (Allentoft et al. 2015). The Yamnaya culture, associated with late Proto-Indo-Europeans, emerged during this time period in the Southwestern Siberian Ural region and the Pontic steppe region of Southeastern Europe (Allentoft et al. 2015; Haak et al. 2015; Jones et al. 2015). Yamnaya steppe herders, who traced their origins to Eastern European and Caucasus hunter-gatherer groups, had largely replaced the Neolithic farming culture in Eastern Europe. As a result, the Bronze Age farmers throughout much of Europe had more hunter-gatherer ancestry compared with their predecessors (Haak et al. 2015). At the same time, early Western and Eastern Europeans came into contact during the Bronze Age. The Late Neolithic Corded Ware people from Germany traced ∼75% of their ancestry to the Yamnaya people (Haak et al. 2015). These migration events influenced the present-day population structures of both Europe and Siberia, but their details remain largely unclear. Little is known about the genetic makeup of Siberian indigenous groups and their genetic links to European and East Asian populations. Furthermore, previous studies have not estimated the genetic contribution of ancient Eurasians to the genomes of modern Siberian indigenous groups, particularly for the Western Siberian populations of Mansi, Khanty, and Nenets people. Siberia is commonly subdivided into western and eastern regions. The territory of Western Siberia extends from the Trans-Uralic region in the west to the Yenisei River in the east. Western Siberian ethnic groups of Mansi, Khanty, and Nenets, as well as many other indigenous people from this region, speak languages that are broadly categorized as Uralic and are further subdivided into Ugric, Finno-Permic, and Samoyedic language groups (Supplemental Fig. S1). Western Siberians Khanty and Mansi, together with 13 million Hungarians in central Europe, are members of the Ugric language group. The language of Northwestern Siberian Nenets people belongs to the Samoyedic branch of Uralic languages, suggesting shared history between Nenets people and other Western Siberian populations. Despite the geographical separation, several European populations also speak languages that are related to Ugric languages spoken by Western Siberians. These populations include Komi, Karelians, Veps, Saami, and Finns, who traditionally occupy areas of Northern and Northeastern Europe. Their languages belong to Permic and Finno-Volgaic language groups and are distantly related to other Ugric languages (Supplemental Fig. S1).  Eastern Siberian groups settled in the Siberian taiga region between the Yenisei River in the west and Sea of Okhotsk in the east. Areas that were historically occupied by Evens, Evenks, and Yakuts account for the majority of the Eastern Siberian territory. Traditional territories of Altayan people are in the very center of Asia at the junction of the Siberian taiga, the steppes of Kazakhstan, and the semi-deserts of Mongolia. Kalmyks, who settled in the lower Volga region in Eastern Europe, migrated to these areas in the 17th century from Dzungaria, a region in northwestern China (Erdeniev 1985). Languages of the Eastern Siberian indigenous populations together with the Kalmyk and Altayan languages are broadly categorized as Altaic and are further subdivided into Mongolic, Tungusic, and Turkic groups (Supplemental Fig. S1).  To further understand the genetic relationships and ancient history of indigenous Northeastern European and Siberian populations, spanning thousands to tens of thousands of years, we performed deep genome sequencing of 28 individuals (with an average sequence coverage depth of 38×), representing 14 major ethnic groups from Northeastern Europe and Siberia, including undersurveyed Western Siberian groups (Table 1), and compared them to 32 publicly available high-coverage modern genomes from 18 populations (Supplemental Table S2; Wong et al. 2013, 2014; Zhou et al. 2013; Jeong et al. 2014; Prufer et al. 2014), two hominin genomes, the Neanderthal (Prufer et al. 2014), and Denisova genomes (Meyer et al. 2012), as well as 46 publicly available ancient genomes from other studies (Supplemental Table S3). |
|
|
|
Post by Admin on Mar 29, 2019 18:21:50 GMT
An overview of population relationships First, we sought to obtain a broad overview of population genetic relationships and to gain insight into the levels of genetic heterogeneity of the populations using principal component analysis (PCA). We combined both publicly available DNA microarray genotyping data from 892 present-day human individuals (Li et al. 2008; The 1000 Genomes Project Consortium 2012; Yunusbayev et al. 2012; Fedorova et al. 2013; Khrunin et al. 2013; Zhou et al. 2013; Raghavan et al. 2014), representing 27 diverse Asian, European, Siberian and Native American populations (Fig. 1B; Supplemental Fig. S2; Supplemental Table S1), and 82 individuals from Mansi (N = 45) and Khanty (N = 37) groups genotyped for the first time, using high-density SNP microarrays. To provide relative placement of samples that were a focus of this study, we projected their sequenced genomes onto the principal components. All the sequenced genomes clustered with genotyped samples from corresponding populations, confirming that the sequenced genomes were good representatives of these groups.  Figure 1. Geographic and genetic affinities of European and Siberian populations. (A) Map of Siberia, Western Russia, and Eastern Europe shows geographic locations of individuals sequenced as part of this work. Colored areas of the map show geographical areas traditionally occupied by corresponding populations (Peoples of Russia 1994). The Ural Mountain range, a natural boundary between Europe and Siberia, is shown as blue peaks. Western Siberian ethnic groups that include Mansi, Khanty, and Nenets people inhabit the vast regions of taiga stretching from the Ural Mountains in Western Siberia to the Yenisei River in the Siberian East. Northeastern European populations, including Komi, Karelians, and Veps, occupy areas to the west of Ural Mountains. The Eastern Siberian populations, including Altayans, Yakuts, Buryats, Evens, and Evenks, occupy the Siberian taiga region between the Yenisei River in the west and Sea of Okhotsk in the east. Kalmyks migrated in the 17th century to the lower Volga region from a region in northwestern China. (B) Principal component analysis (PCA) of 892 modern-day humans from 27 populations mainly from Siberia and Europe. PCA was performed using a common set of 137,637 SNPs. Sequenced samples are shown using an asterisk. Samples genotyped using DNA microarrays are shown with dots. The PCA plot captured major differentiation of populations along the first principal component, accounting for 6.4% of the genetic variation. The first principal component corresponds to the west-to-east gradient across Eurasia. The second principal component captured the spread of populations along the north-to-south latitudinal cline, especially among Siberians, capturing 1% of the genetic variation. The PCA plot reveals substantial genetic differentiation between Siberian groups. This suggests that Siberian populations harbor significant genetic diversity not represented by other populations (e.g., European populations, which show only very modest levels of population-specific variation). Admixture events between Western and Eastern Siberians Next, we used TreeMix (Pickrell and Pritchard 2012) to construct a tree-based model of population genetic relationships and inter-population admixtures using a set of 30.1 million genome-wide autosomal SNPs inferred from the whole-genome sequencing data. The model placed European and Asian populations along the two early diverging branches (Fig. 2). Furthermore, the Western Siberian Mansi, Khanty, and Nenets groups formed an early diverging subclade with affinity to other Eastern European populations. Most Eastern Siberians, including the Even, Evenk, Buryat, and Yakut groups, formed a separate lineage more related to East Asian populations.  Figure 2. Genetic relationships of modern-day Siberian, Asian, Native American, European, and African populations. Autosomal TreeMix admixture graph based on whole-genome sequencing data from 44 individuals primarily from Africa, Europe, Siberia, and America. The x-axis represents genetic drift, which is proportional to the effective population size Ne. Admixture events are shown with arrows, with admixture intensities indicated by percentages. Residuals are shown in Supplemental Figure S5. The TreeMix model also revealed several important admixture events. In particular, we found that 43% (95% CI: 38%–47%) of the Western Siberian ancestry could be attributed to an ancient admixture, with the Eastern Siberian population most related to the modern-day Evenk people. The Northwestern Siberian Nenets people exhibited evidence of an additional admixture, with the Eastern Siberian population closely related to modern-day Evens. We estimated that 38% (95% CI: 31%–46%) of the Nenets’ ancestry could be attributed to this admixture event. To further confirm these predictions, we computed D-statistics (Durand et al. 2011; Patterson et al. 2012) to test for an excess of shared alleles between the Western Siberian and Eastern Siberian groups (Supplemental Figs. S7A–C, S8A–C, S18). In agreement with the TreeMix inference, we observed significant genetic affinity between the Mansi and Evenk, the Khanty and Evenk, and the Nenets and Even people based on the D-statistics tests (Supplemental Figs. S7A–C, 8A–C). These observations support the ancient admixture between ancestors of modern Western and Eastern Siberian populations. Notably, TreeMix also inferred an admixture signal between common ancestors of the Mansi, Khanty, and Nenets and Native American Andean Highlanders, an indication of their shared genetic history. This event accounts for 6% (95% CI: 4%–8%) of the Western Siberian ancestry. As discussed below, this admixture can be explained by a genetic link of both Western Siberians and Native Americans to ANEs. Phylogenetic trees provide simplified demographic models, lacking precise information about divergence times of ancestral lineages. The lengths of TreeMix branches are proportional to units of drift parameter (Pickrell and Pritchard 2012) and cannot be directly related to the time of population divergence. Furthermore, branch lengths can be affected by population bottlenecks, which further complicates the inference of population divergence time and limits interpretability of the model. In an attempt to provide more direct estimates of the divergence time between populations, we employed multiple sequentially Markovian coalescent (MSMC) analysis (Fig. 3A; Schiffels and Durbin 2014). We designated the time at which the relevant cross-coalescence rate between two populations becomes 0.5 as a proxy for their separation time (Fig. 3B). The MSMC method is particularly informative for events that are older than 10,000 yr, when each population is represented by only a single individual (Schiffels and Durbin 2014); estimates more recent than 10,000 yr ago are less precise, unless more individuals are included in the analysis.  Figure 3. MSMC analysis of population separation times. (A) Separation times were calculated for each of the five reference individuals (French, Andreapol Russian, Mansi, Evenk, and Han). Reference populations are shown at the top (inside the orange rounded rectangles). Each reference population column shows MSMC separation time estimates between the reference population and other populations of interest. Populations of interest were sorted according to their separation time from diverging most recently (top) to the oldest (bottom). Separation times are shown on the left side of each graph. Populations with similar times were grouped together. (And) Andreapol; (Ust) Ustyuzhna; (Ob) Objachevo. MSMC separation times under 10,000 yr may be noisy, and therefore, such separation times and corresponding populations are shown in a gray-colored font. (B) MSMC cross-coalescence plots corresponding to divergence between Mansi and five other populations (Nenets, Evenks, French, Andean Highlanders, Dinka). The median separation value (y = 0.5; orange line) was used as a proxy for the population divergence time. Our MSMC analysis suggested that the Mansi, Khanty, and Nenets separated relatively recently (4.8 kya), after the ancestral populations of Western Siberians experienced an admixture with an ancient group most related to the modern-day Evenk population (Fig. 2). This admixture can be dated to ∼6.8 kya based on the estimated separation time between the Mansi and Evenk populations. These time estimates are only approximate and should be interpreted with caution; however, the relative order of populations (Fig. 3A) is still informative for interpreting their relative genetic affinities. To further confirm these estimates, we performed a more computationally intensive, but more accurate eight-haplotype MSMC analysis using two individuals per population. The results from this analysis provided a similar estimate of 9.9 kya for the time of the Eastern Siberian admixture into Western Siberian populations (Supplemental Fig. S9). Therefore, the Eastern and Western Siberian populations likely came into contact within the last 5000–10,000 yr. |
|
|
|
Post by Admin on Mar 30, 2019 18:14:23 GMT
Relationships of modern-day Europeans and Siberians The phylogenetic tree model placed almost all modern-day Europeans (except for Kalmyks, who migrated to Europe ∼400 yr ago) along a single branch, with the divergence pattern mimicking the east-to-west distribution of populations within Europe (Fig. 2). Confirming the observation from the PCA analysis (Fig. 1B) that Western and Eastern European populations were fairly close genetically, MSMC results showed that the separation times between the French and Eastern European populations fell in the range of 2.2 kya (French-Karelian) to 7 kya (French-Komi Objachevo). In contrast, the smallest separation time estimate between the French and Siberian populations was 12.5 kya (French-Khanty), exceeding the separation time between any two European populations by a wide margin. To provide additional support for our autosomal results, we analyzed SNPs within a portion of Y-Chromosome and estimated divergence times of population-specific haplogroups (Fig. 4A). Our Y-Chromosome analysis (Methods; section “Y-Chromosome and mtDNA Analyses” in Supplemental Information) showed evidence of gene flow from the Siberian populations into Eastern and Northern Europe. A Y-Chromosome clade, the N haplogroup, was linked to ancient Siberian populations (Fu et al. 2014). The N haplogroup is common in both modern Siberia and Eastern Europe (Malyarchuk et al. 2004; Lappalainen et al. 2008; Mirabal et al. 2009) but not among Western Europeans. We observed an expansion of N1c1 Y-Chromosome haplogroup among Siberian (Evens, Evenks, Mansi, Khanty) and Northeastern European populations (Veps and Komi) that occurred ∼5.3–7.1 kya. This suggests that the N1c1 haplogroup reached Europe only relatively recently and had limited spread among populations of Western Europe.  Figure 4. Y-Chromosome and mtDNA phylogeny and haplogroup divergence times. (A) Y-Chromosome tree topology was calculated using maximum likelihood approach implemented in MEGA tool (Tamura et al. 2011) using 12,394 polymorphic sites within high-confidence regions defined by Poznik et al. (2013). The x-axis represents divergence time of haplogroups and is expressed in units of thousands of years ago (1 kya = 1000 yr ago). The tree was constructed using the data from 54 modern-day humans from 30 populations. Split times were calculated according to the number of derived alleles assigned to each branch (Methods), and splits were placed along the x-axis to reflect haplotype divergence time. Terminal branches end at X = 0 for modern-day individuals. Seven ancient individuals were placed on the haplotype tree according to the SNP-sharing patterns with high-confidence–derived allele SNVs assigned to each branch. Branches of ancient individuals (orange lines) terminate at times when each ancient individual lived. Haplogroups were assigned based on ISOGG Y-Chromosome SNP index and shown along the right side of the tree (International Society of Genetic Genealogy 2014). Individuals sequenced as part of this work are shown in red. (B) mtDNA phylogeny was constructed using MEGA tool in a similar fashion to the Y-Chromosome tree. The tree includes data from 70 individuals and 34 populations, as well as eight ancient genomes. (C) Divergence times of haplogroups from Y-Chromosome phylogeny (left) and mtDNA phylogeny (right). Diverging clades are shown on the left sides (“Split” columns). Confidence intervals were calculated using Poisson model. Interestingly, the tree constructed from mtDNA haplotypes (Fig. 4B) did not show a similarly distinct clade shared among the Siberians and Northeastern Europeans that diverged 5–10 kya. We note, however, that the mtDNA tree has a greater diversity of haplogroups, which are sampled very sparsely across individuals that we sequenced. We further used D-statistics test to test for gene flows occurring during the last 7000 yr between the Western Siberian and Northeastern European populations (Fig. 5A,B; Supplemental Figs. S7G,K,L, S8G,K,L, S13A,B). In agreement with the N1c1 haplogroup expansion, Northeastern Europeans, including Mezen Russians, Veps, Karelians, and Komi, showed statistically significant admixture signals with Siberian groups such as the Nenets, Evens, Yakut, and Khanty. These admixtures were much weaker among more southern populations in Eastern Europe, such as Ustyuzhna and Andreapol Russians, as well as a Belarusian individual (Supplemental Figs. S7H–J, S8H–J), indicating that Siberian admixtures are particularly strong among Northeastern Europeans.  Figure 5. Analysis of admixtures using D-statistics. Genetic affinities were determined using D-statistics test D(P1, P2, P3, O) as defined previously (Durand et al. 2011). (A) Mezen Russian affinities; (B) Veps affinities; (C) Ust’-Ishim affinities; (D) Mal'ta boy (MA-1) and Afontova Gora (AG-2) affinities; (E) Ire8 and Ajv52 affinities. The affinities of a variable population X (orange color) relative to other populations were calculated using ADMIXTOOLS (Patterson et al. 2012) and shown under the D column. Statistical significance of admixture was expressed as a z-score and shown under the Z column. A green font was used to highlight significant admixtures between P1 and P3 (Z > 3). A blue font was used to highlight significant admixture between P2 and P3 (Z < −3). African San was used as an outgroup in all tests. (And) Andreapol; (Ust) Ustyuzhna; (Ob) Objachevo. D-Statistics results using different outgroups (Chimp, Mandenka, Mbuti) are shown in Supplemental Figures S13 and S17. Genetic affinity between Eastern Siberians and East Asians Based on the TreeMix results, we observed that Eastern Siberians had a stronger genetic affinity to Native American and East Asian populations than to Europeans (Fig. 2). Eastern Siberian populations, such as Buryats, Yakuts, Evens, and Evenks, form a distinct sublineage after diverging from Han, Dai, and Sherpa, the East Asian populations (Fig. 2; Supplemental Figs. S3, S4). By use of MSMC, we estimated the separation time between Eastern Siberians and East Asian populations was between 8.8–11.2 kya (based on Evenk-Han and Evenk-Sherpa separation time estimates) (Fig. 3A). The existence of the Eastern Siberian autosomal clade, and its affinity to East Asian populations, was well supported by multiple TreeMix models (Figs. 2, 6, 7; Supplemental Fig. S3), as well as 100% of the bootstrap replicates (Fig. 6). Thus, our results support the separation of Eastern Siberian lineage from other East Asian groups, and we date this separation event to ∼10 kya.  Figure 6. Genetic relationships of Ust’-Ishim and modern-day humans. TreeMix model with seven admixture events incorporates ancient Siberian Ust’-Ishim. Blue circles indicate branching events that were investigated using 100 bootstraps, and percentages of trees supporting the branch point are shown. Residuals are shown in Supplemental Figure S14. Kalmyks originated from northwestern China but later migrated to Southeastern Europe; thus, it would be illuminating to determine their genetic affinities to other populations, especially to other Siberian groups. To clarify the relationship of Kalmyks and their historical geographical neighbors in Siberia and East Asia, we performed additional D-statistics analysis. Both the Kalmyks and Altayans were moderately closer to other Eastern Siberians, including the Evens and Evenks (D-statistics = 0.13–0.146; Supplemental Figs. S7D,E, S8D,E), than to East Asians (D statistics = 0.11–0.125). However, unlike Kalmyks, Altayans additionally traced ∼37% (95% CI: 31%–43%) of their ancestry to another unknown population, which the model predicted to be related both to modern Europeans and Western Siberians (Fig. 2). |
|

