|
|
Post by Admin on Apr 10, 2019 18:36:10 GMT
Neanderthal ancestry We next analyzed Neanderthal admixture contributions to the ancestry of Q1 (Bedouin) compared to the Q2 (Persian-South Asian) and Q3 (African) Qataris, the 1000 Genomes populations, and the populations of the Human Origins samples using the F4 ratio and Patterson's D-statistic (Fig. 4; Supplemental Fig. 10, Supplemental Table XI; Patterson et al. 2012). The results for both methods were highly correlated (Supplemental Fig. 10A). The Q1 (Bedouin; F4 ratio = 0.026, D-statistic = 0.000) had more Neanderthal admixture than all African populations, including Q3 (African; F4 ratio range = −0.017 to 0.024, D-statistic range = −0.031 to −0.003). The Q1 (Bedouin) also had Neanderthal admixture at levels comparable to Q2 (Persian-South Asian; F4 ratio = 0.024, D-statistic = −0.003) and to other Middle Eastern populations, including other Bedouin populations (Human Origins Bedouin A F4 ratio = 0.022, D-statistic = −0.003 and Bedouin B F4 ratio = 0.024, D-statistic = −0.003) and Saudi (F4 ratio = 0.026, D-statistic = −0.001). Interestingly, the Q1 (Bedouin) did not tend to have higher Neanderthal admixture levels when considering populations outside of the Middle East, where the bulk of European populations had higher Neanderthal admixture (F4 ratio range = 0.018 to 0.041, D-statistic range = 0.003 to 0.010). Yet, the percentage of Neandethal admixture with the Q1 (Bedouin) was higher than expected if it could be entirely explained by later admixture events between the Q1 (Bedouin) and Europeans (observed F4 ratio = 0.026 versus expected F4 ratio = 0.00247).  Figure 4. The higher Neanderthal ancestry in the Q1 (Bedouin) Qatari compared to African populations places the divergence of ancestral Arabs after the out-of-Africa bottleneck. Given the current evidence of the geographic range of Neanderthal populations stretching from Europe and the Mediterranean through Northern and Central Asia (Fu et al. 2014; Hershkovitz et al. 2015), the lower Neanderthal Ancestry in the Q1 (Bedouin) Qatari compared to populations within the ancestral Neanderthal range is also consistent with an early divergence of the ancestors of indigenous Arabs from other lineages that populated Asia and Europe. Yet, since the Neanderthal admixture in the Q1 (Bedouin) cannot be entirely explained by admixture with Europeans, this indicates there was some admixture between Neanderthals and ancestors of the Q1 (Bedouin) in the region of the Arabian Peninsula. TreeMix analysis We also analyzed the autosomes of the combined 96 Q1 (Bedouin), Q2 (Perisan-South Asian) or Q3 (African) Qataris, and non-admixed populations of the 1000 Genomes Project using the population split and mixture inference method TreeMix (Pickrell and Pritchard 2012) to assess the relative genetic similarity of populations based on high-density, genome-wide allele frequencies. The analysis returned an overall tree for the 1000 Genomes populations that mirrored those found previously (Shriner et al. 2014) with the addition of the Q1 (Bedouin) and Q2 (Persian-South Asian) clustering on the branch that includes Europeans (Pérez-Miranda et al. 2006) and the Q3 (African) clustering with African populations (Fig. 5). When migrations were allowed in the analysis, no migration events were observed between the Q1 (Bedouin) and African populations, even when allowing as many as five migration events (Supplemental Fig. 11). These results are also consistent with what is known of the migration history of the Arabian Peninsula, including migration both to and from Europe during ancient and more recent eras of civilization, where this resulted in detectable admixture from European populations in both the Q1 (Bedouin) and Q2 (Persian-South Asian) (Omberg et al. 2012).  Figure 5. TreeMix (Pickrell and Pritchard 2012) hierarchical clustering analysis of the Q1 (Bedouin), Q2 (Persian-South Asian), and Q3 (African) and the 1000 Genomes Project samples. Shown is a maximum-likelihood tree of population splits inferred without subsequent migration events, in which branch lengths estimate divergence between populations (Europeans in shades of purple: CEU, FIN, GBR, IBS, TSI; East Asians in shades of brown: CHB, CHS, JPT; Africans in shades of orange: LWK, YRI, with the Q1 [Bedouin] in red, Q2 [Persian-South Asian] in azure, and Q3 [African] in black). When allowing from one to five migration events in separate TreeMix analyses, none of the admixture loops connected the Q1 (Bedouin) with any African populations (Supplemental Fig. 10), consistent with the Q1 (Bedouin) having no recent African admixture. Proportion of shared alleles neighbor-joining analysis As the principal component analysis and the TreeMix population-level clusterings depend on allele frequencies, the clustering of the Q1 (Bedouin) on a common branch with European populations could be driven by the haplotypes introduced by migrants, which would be expected to shift the allele frequencies of these populations toward each other. As such, these clusterings based on allele frequencies do not necessarily argue against significant and deep ancestry of the Q1 (Bedouin) on the Arabian Peninsula, as indicated by the levels of Neanderthal admixture in this subpopulation. Additionally, these population-level clusterings are disproportionately influenced by common segregating alleles (Pickrell and Pritchard 2012), while rare alleles can be more informative about deeper shared ancestry (Mathieson and McVean 2014) as the identity by state of a rare variant can more accurately reflect identity by descent (Hochreiter 2013).  Figure 6. In contrast to population-level clustering, a pairwise clustering of individual genomes based on shared variants provides a relative measure for comparing total shared ancestry between individuals. Also, when applied to a common set of genome-wide, high-density markers that include the low-minor allele frequency alleles of the 1000 Genomes Project, such pairwise clustering also provides an appropriate weight to rare alleles. We therefore performed a proportion of shared alleles (Mountain and Cavalli-Sforza 1997) analysis on the combined samples in the 104 Qatari and the 1000 Genomes samples, in which pairwise proportion of shared alleles was calculated for the 11,711,386 autosomal, biallelic SNPs segregating in both the 104 Qatari and the 1000 Genomes samples. A robust version of the neighbor-joining algorithm was used to perform a pairwise clustering of the samples (Fig. 6A–F; Criscuolo and Gascuel 2008), in which bootstrap support values were calculated for the observed trees using 100 random samplings of the SNPs. The neighbor-joining analysis revealed that 50 of the 56 Q1 (Bedouin), along with three Q2 (Persian-South Asian), one Q3 (African), and two Q0 (Subpopulation Unassigned) Qataris, clustered outside African lineages and were also the most extreme outgroup that are basal to all non-African populations lacking recent African admixture (Fig. 6D). Strong bootstrap support was observed for this cluster (70 of 100 iterations), and for its presence as an outgroup to the Eurasian cluster (68 of 100 iterations), comparable to the support for the Japanese cluster (60 of 100 iterations) and for the East Asians as an outgroup to Europeans and Americans (81 of 100 iterations). The Q1 (Bedouin) therefore fit the criteria of having ancient migration from Africa and being most distantly related to all other non-Africans in total ancestry. A total of 11 Q2 (Persian-South Asian), three Q1 (Bedouin), one Q3 (African), and one Q0 (Subpopulation Unassigned) defined an Asian outgroup more closely related to Asians than the main Q1 (Bedouin) outgroup (Fig. 6C), likely driven by the ancestry of the the Q2 (Persian-South Asian) subpopulation traceable to Persia and South Asia (Omberg et al. 2012) and indicating these individuals are most distantly related to other Asians present in this cluster. A total of 12 Q3 (African), three Q1 (Bedouin), three Q2 (Persian-South Asian), and four Q0 (Subpopulation Unassigned) cluster as long individual branches or small clusters between the major Q1 (Bedouin) cluster and the admixed individuals of African ancestry from Southwest US (ASW), potentially representing individuals with a higher proportion of African admixture. As expected from the analyses of population genetic similarity and prior neighbor-joining analysis of admixed populations (Kopelman et al. 2013), the Q3 (African) and African Americans do not form large clusters, but rather appear as multiple individual branches close to the indigenous African populations, most similar to their African admixture source (Fig. 6E,F). A set of three Q2 (Persian-South Asian) clustered as an outgroup to the Tuscan Southern European (TSI) branch (Fig. 6B), which is not unexpected given admixture with European populations (Omberg et al. 2012; Pickrell et al. 2014). |
|
|
|
Post by Admin on Apr 11, 2019 17:56:56 GMT
The hypothesis that the first Eurasian populations were established on the Arabian Peninsula and that contemporary indigenous Arabs are direct descendants of this ancient population is supported by two major conclusions derived from the combined evidence of this study. First, the analysis results for X/A diversity, the pairwise sequential Markov coalescent, genome-wide admixture, timing of African admixture, local admixture deconvolution, Neanderthal admixture, and application of TreeMix, support the inference that the Q1 (Bedouin) can trace the bulk of their ancestry back to the out-of-Africa migration events. Second, the combination of lower levels of Neanderthal admixture in the Q1 (Bedouin) than European/Asian populations and the outgroup position of the Q1 (Bedouin) compared to non-Africans in the pairwaise similarity clustering of high-density variants measured genome-wide, place the Q1 (Bedouin) as being the most distant relatives of other contemporary non-Africans. Given that the Q1 (Bedouin) have the greatest proportion of Arab genetic ancestry measured in contemporary populations (Hodgson et al. 2014; Shriner et al. 2014) and are among the best genetic representatives of the autochthonous population on the Arabian Peninsula, these two conclusions therefore point to the Bedouins being direct descendants of the earliest split after the out-of-Africa migration events that established a basal Eurasian population (Lazaridis et al. 2014). This is also consistent with the majority of Q1 (Bedouin) being able to trace a significant portion of their autosomal ancestry through lineages that never left the peninsula after the out-of-Africa migration events since such deep ancestry would not be expected if the entire Arabian Peninsula population had been reestablished from Africa or a non-African population at a later point.  Given the complex history of migration patterns to and from European populations, and the complicated patterns of isolation and intra- and inter-marriage of the indigenous Bedouin populations (Hunter-Zinck et al. 2010; Sandridge et al. 2010), it is not surprising that among the Q1 (Bedouin) are individuals who retain an autosomal signal of being the most distant relatives of non-Africans, while population-level clustering based on migration-shifted allele frequencies places the Q1 (Bedouin) closer to Europeans. The basal position of the Q1 (Bedouin) also has interesting implications for theories about the frequency, timing, and path of major migration waves that established populations in Asia and Europe (Shi et al. 2008; Lazaridis et al. 2014; Shriner et al. 2014). A few isolated Asian populations were previously suspected to be descendants of a separate out-of-Africa migration wave based on Y Chromosome data (Hammer et al. 1998; Shi et al. 2008). Yet, distinct out-of-Africa migration events or separate migration waves emanating from the Arabian Peninsula into Europe and West Asia would be expected to place Bedouins/Europeans and Asians on separate branches of a pairwise clustering tree, distinct from our finding that places the Q1 (Bedouin) as direct descendants of the earliest lineage that split from the ancient non-African population. 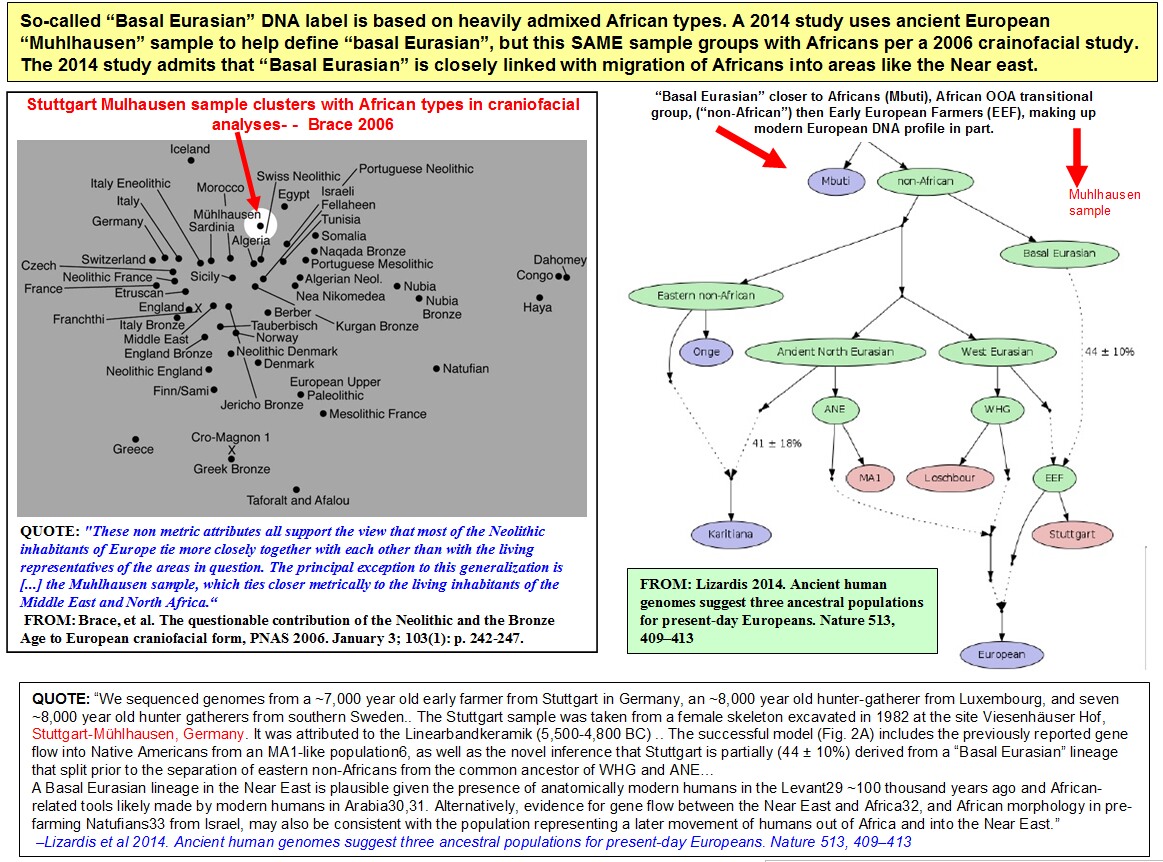 A demographic scenario consistent with the evidence presented here is that the population ancestral to the Q1 (Bedouin) migrated out of Africa, and a subset of this population remained in the peninsula until the present day, while a second subset of this population migrated onward and colonized Eurasia. This migration scenario implies the signal of the same bottleneck would be present in all non-African populations, which has been observed thus far in coalescent analysis of contemporary non-African populations (Gronau et al. 2011; Fu et al. 2014; Schiffels and Durbin 2014) and for an anatomically modern human who lived 45,000 yr ago (Fu et al. 2014). This is also consistent with the recent discovery of another anatomically modern human who lived 55,000 yr ago just northeast of the Arabian Peninsula that had morphological features similar to European peoples (Hershkovitz et al. 2015), where this individual could have been a descendant of the basal Eurasian population that remained on the peninsula. Under this migration scenario, although other waves of migration may have occurred, the descendants of these alternative waves either left no descendants or were integrated into the dominant populations.  Beyond the importance for disentangling human migration history, an early split of Eurasian lineages in the Arabian Peninsula has implications for the study of disease genetics for indigenous people in the region. For example, for a disease such as type 2 diabetes that has a prevalence of >18% in the Qatari population, associated genetic variants would not a priori be expected to be the same as those discovered in Europeans, when considering that indigenous Arabs are able to trace a significant portion of their ancestry back to ancient lineages on the Arabian Peninsula. More generally, this suggests that for any genome-wide association study (GWAS) or rare variant association study (RVAS) of diabetes or other complex diseases in Qatar, inference of deep ancestry in the Arabian Peninsula, using rare variation sampled by genome or exome sequencing, is critical for identifying new disease risk genes. Given the dearth of next generation sequencing studies conducted in Middle Eastern and Arab populations, these results indicate that a considerable number of variants that make important contributions to disease risk in these populations are yet to be discovered. This study is the first analysis of Arabian Peninsula migration making use of deeply sequenced genomes from a sample of unrelated inhabitants of the peninsula. Although there have been many analyses of Chr Y and mtDNA sampled from Arab individuals (Abu-Amero et al. 2007, 2008, 2009; Rowold et al. 2007), and there have been previous surveys of genetic variation of people within the peninsula and immediately surrounding regions conducted with genotyping arrays (Behar et al. 2010; Hunter-Zinck et al. 2010; Alsmadi et al. 2013; Markus et al. 2014; Shriner et al. 2014) and deep exome sequencing (Rodriguez-Flores et al. 2012, 2014; Alsmadi et al. 2014), and by individual high-coverage genomes (Alsmadi et al. 2014; John et al. 2015), the sample of rare and common genetic variation throughout the genome in our sample provides a far more complete picture of how both ancient and recent migration events have contributed to the genetics of the modern peoples of the Arabian Peninsula. For understanding how human migration history has determined the structure of modern genomes, our identification of a cluster of Q1 (Bedouin) as the most distant ancestors of non-Africans is of considerable interest, particularly given the suspected route of migration out of Africa and into the surrounding continents. The possibility that the Q1 (Bedouin) are descendants of the first Eurasians provides an additional piece of the puzzle concerning ancient migration routes and the establishment of ancient non-African populations. Genome Res. 2016. 26: 151-162 |
|
|
|
Post by Admin on Apr 21, 2019 17:44:42 GMT
nterbreeding between Neandertals and modern humans ∼55,000 y ago has resulted in all present-day non-Africans inheriting at least 1–2% of their genomes from Neandertal ancestors (1, 2). There is significant heterogeneity in the distribution of this Neandertal DNA across the genomes of present-day people (3, 4), including a reduction in Neandertal alleles in conserved genomic regions (3). This has been interpreted as evidence that some Neandertal alleles were deleterious for modern humans and were subject to negative selection following introgression (3, 5). Several studies have suggested that low effective population sizes (Ne) in Neandertals led to decreased efficacy of purifying selection and the accumulation of weakly deleterious variants. Following introgression, these deleterious alleles, along with linked neutral Neandertal alleles, would have been subjected to more efficient purifying selection in the larger modern human population (6, 7). In apparent agreement with this hypothesis, a study of Neandertal ancestry in a set of anatomically modern humans from Upper-Paleolithic Europe used two independent statistics to conclude that the amount of Neandertal DNA in modern human genomes decreased monotonically over the last 45,000 y (Fig. 1A, dashed line) (8). This decline was interpreted as direct evidence for continuous negative selection against Neandertal alleles in modern humans (8⇓⇓–11). However, it was not formally shown that selection on deleterious introgressed variants could produce a decline in Neandertal ancestry of the observed magnitude. Nevertheless, this decrease in Neandertal ancestry—together with the suggestion of a higher burden of deleterious alleles in Neandertals—are now commonly invoked to explain the fate of Neandertal ancestry in modern humans (9⇓⇓–12).  Fig. 1. Direct and indirect f4-ratio estimates of Neandertal ancestry. (A) Best linear fits for indirect and direct f4-ratio estimates of Neandertal ancestry in ancient and modern West Eurasians (solid points for direct f4-ratio, “x” for indirect f4-ratio). Shaded areas are 95% CIs (SI Appendix, section S1). (B) Tree model and formula used for the indirect f4-ratio. (C) Tree model and formula used for the direct f4-ratio. Present-day individuals are West Eurasians from the SGDP panel, excluding individuals from the Near East (Neandertal ancestry for all West Eurasians shown in SI Appendix, Fig. S7). Here, we reexamine estimates of Neandertal ancestry in ancient and present-day modern humans, taking advantage of a second high-coverage Neandertal genome that recently became available (13). This allows us to avoid some key assumptions about modern human demography that were made in previous studies. Our analysis shows that the Neandertal ancestry proportion in Europeans has not decreased significantly over the last 45,000 y. Using simulations of selection and introgression, we show that a model of weak selection against deleterious Neandertal variation also does not predict significant changes in Neandertal ancestry during the time period covered by existing ancient modern human samples. In contrast, these simulations do predict a depletion of Neandertal ancestry around functional genomic regions. We then use our updated Neandertal ancestry estimates to examine the genomic distribution of introgressed Neandertal DNA and find that selection against introgression was strongest in regulatory and conserved noncoding regions compared with protein-coding sequence (CDS), suggesting that regulatory differences between Neandertals and modern humans may have been more extreme than protein-coding differences. Previous Neandertal Ancestry Estimate. A number of methods have been developed to quantify Neandertal ancestry in modern human genomes (14). Among the most widely used is the f4-ratio statistic, which measures the fraction of drift shared with one of two parental lineages to determine the proportion of ancestry, α, contributed by that lineage (Fig. 1 and SI Appendix, Fig. S1) (15, 16). Although they have been used to draw inferences about gene flow between archaic and modern human populations, f4-ratio statistics are known to be sensitive to violations of the underlying population model (15). Estimating α, the proportion of ancestry in X contributed by a lineage A, requires a sister lineage B to lineage A which does not share drift with X after separation of B from A (SI Appendix, Fig. S1). Fu et al. (8) used an f4-ratio statistic to infer the contribution from an archaic lineage by first estimating the proportion of East African ancestry in a non-African individual X, under the assumption that Central and West Africans (B) are an outgroup to the East African lineage (A) and to the modern human ancestry in non-Africans. Defining this East African ancestry proportion as α = f4(C. and W. Africans, Chimp; X, Archaics)/f4(C. and W. Africans, Chimp; E. Africans, Archaics), the proportion of archaic ancestry was then calculated simply as 1 − α, under the assumption that all ancestry that is not of East African origin must come from an archaic lineage (8). We refer to this statistic as an “indirect f4-ratio.” Given the sensitivity of the f4-ratio method to violations of the underlying population models (15), we explored the validity of assumptions on which this calculation was based. In addition to the topology of the demographic tree, which has recently been shown to be incorrect (17), the indirect f4-ratio assumes that the relationship between Africans and West Eurasians has remained constant over time (8). However, our understanding of modern human history and demography have been challenged by new fossil discoveries (18) and the analysis of ancient DNA, with several studies documenting previously unknown migration events in both West Eurasia (19) and Africa (17, 20, 21). Furthermore, an f4 statistic sensitive to changes in the relationships between West Eurasians and various African populations [formulated as f4(Ust’-Ishim, X; African, Chimp), where X is a West Eurasian individual] shows increasing allele sharing between West Eurasians and Africans over time (SI Appendix, Fig. S2A). In contrast, f4(Ust’-Ishim, Papuan; African, Chimp) is not significantly different from zero (|Z| < 1 when using Dinka, Yoruba, or Mbuti in the third position of the f4 statistic), demonstrating that this trend is not shared by all non-Africans.  Fig. 2. Neandertal ancestry estimates in neutral simulations of migration. Genomic data were simulated under a base model of 3% Neandertal admixture, Ne = 6,000 in Europeans and Ne = 14,000 in two African populations (SI Appendix, Fig. S8, section S2). (A–C) The effect of three migration parameters on direct and indirect f4-ratio estimates of Neandertal ancestry (dotted and solid colored lines, respectively). To evaluate the sensitivity of the indirect f4-ratio to migration events, we performed neutral simulations of Neandertal, West Eurasian, and African demographic histories (Fig. 2). All simulations included introgression from Neandertals into West Eurasians, and varying levels of migration between Africans and West Eurasians, and between African populations. We find that gene flow from West Eurasians into Africans leads to misestimates of Neandertal ancestry when using the indirect f4-ratio statistic, and results in the incorrect inference of a continuous decline in Neandertal ancestry. This decline is not observed in the true simulated Neandertal ancestry (Fig. 2A). The magnitude of this bias depends on the total amount of West Eurasian gene flow into Africa, with larger amounts leading to apparent steeper declines (Fig. 2A). Additionally, gene flow between the two African populations used in the indirect f4-ratio calculation leads to overestimation of the true level of Neandertal ancestry (Fig. 2C). Overall, we find that a combination of West Eurasian migration to Africa and gene flow between African populations can produce patterns that are very similar to those observed in the empirical data (Fig. 2D and SI Appendix, Fig. S3A). However, we caution that effective population sizes and the timing of migration also affect these estimates (SI Appendix, Fig. S3), and that there are likely many additional models that match the empirical data. We note that an independent statistic, using a different set of genomic sites in the same ancient individuals, had been used as a second line of evidence for an ongoing decrease in Neandertal ancestry (8). This statistic, which we refer to as the “admixture array statistic,” measures the proportion of Neandertal-like alleles in a given sample at sites where present-day Yoruba individuals carry a nearly fixed allele that differs from homozygous sites in the Altai Neandertal (22). Much like the indirect f4-ratio, we find that the admixture array statistic is affected by gene flow from non-Africans into Africans and incorrectly infers a decline in the Neandertal ancestry over time (Fig. 2D). |
|
|
|
Post by Admin on Apr 22, 2019 17:43:02 GMT
A Robust Statistic to Estimate Neandertal Ancestry. The recent availability of a second high-coverage Neandertal genome allows us to estimate Neandertal ancestry using two Neandertals—an individual from the Altai Mountains, the so-called “Altai Neandertal” (23) and an individual from the Vindija Cave in Croatia, the so-called “Vindija Neandertal” (13). Specifically, we can estimate the proportion of ancestry coming from the Vindija lineage into a modern human (X) using the Altai Neandertal as a second Neandertal in an f4-ratio calculated as f4(Altai, Chimp; X, African)/f4(Altai, Chimp; Vindija, African), which we refer to as a “direct f4-ratio” (Fig. 1C and SI Appendix, Fig. S1). Note that unlike the indirect f4-ratio described previously, the f4-ratio in this formulation does not make assumptions about deep relationships between modern human populations (Fig. 1C and SI Appendix, Fig. S1). Instead, it assumes that any Neandertal population that contributed ancestry to X formed a clade with the Vindija Neandertal. Recent analyses showed that this is the case for all non-African populations studied to date, including the ancient modern humans in this study (13, 24). When calculated on the simulations described above, we find that the direct f4-ratio is more robust than the indirect f4-ratio (Fig. 2). In fact, its temporal trajectory always closely matches the true simulated Neandertal ancestry trajectory, regardless of the specific parameters of gene flow between non-Africans and Africans (Fig. 2). We note that gene flow from West Eurasians into Africans, which introduces introgressed Neandertal alleles into Africa, produces a slight underestimate of Neandertal ancestry in all samples (Fig. 2A). This is in agreement with empirical direct f4-ratio estimates, which vary depending on the African population used in the calculation, with African populations known to carry West Eurasian ancestry (e.g., Mozabite, Saharawi) (17, 25) generating the lowest estimates (SI Appendix, Fig. S4). Crucially, when we use the direct f4-ratio to estimate the trajectory of Neandertal ancestry in ancient and present-day Europeans, we observe nearly constant levels of Neandertal ancestry over time (Fig. 1A, points and solid line) and find that a null model of zero slope can no longer be rejected (Fig. 1A, P = 0.36, estimated via resampling as described in SI Appendix, section S1). We note that these estimates are based on a relatively small number of individuals, especially for older time points, and that the CIs are wide. For example, we cannot reject a linear decline in Neandertal ancestry of approximately half a percent over the timespan of this dataset (95% CI −0.51–0.37%). Additionally, these analyses are performed on SNPs that were ascertained largely in present day individuals. To examine the effects of such ascertainment, we split the dataset based on the ascertainments used and recalculated the direct and indirect f4-ratios on each of the subsets (SI Appendix, Fig. S5). Although the slopes show some variability, in all but one ascertainment subset the direct f4-ratio cannot reject a slope of 0, whereas the indirect f4-ratio consistently rejects a slope of 0, suggesting that these results are robust to the effects of ascertainment (SI Appendix, Fig. S5). In addition to calculating direct f4-ratio estimates, we estimated Neandertal ancestry proportions using the qpAdm method (26) and obtained similar results (null model of zero slope using Neandertal ancestry point estimates cannot be rejected with P = 0.17). Our observation that there has been no change in Neandertal ancestry over the past 45,000 y has several implications for our understanding of the fate of Neandertal DNA in modern humans. First, it constrains the timescale during which selection could have significantly affected the average genome-wide Neandertal ancestry in modern humans, an issue addressed below in more detail. Second, a previous analysis of a 40 ky old individual (“Tianyuan”) from East Asia applied the indirect f4-ratio statistic to estimate his Neandertal ancestry proportion at 5% (27). When we apply the direct f4-ratio statistic for this individual, we arrive at a value of ∼2.1% (using Dinka as the African group in the calculation). Third, it has consequences for the so-called “dilution” hypothesis, which suggests that lower levels of Neandertal ancestry in Europeans compared with East Asians can be explained by dilution of Neandertal ancestry in Europeans due to admixture with a hypothetical Basal Eurasian population that carried little to no Neandertal ancestry (19, 28). Previous studies have found Basal Eurasian ancestry in all modern and some ancient Europeans [in this study, four ancient individuals show evidence of Basal Eurasian ancestry: Satsurblia (15 kya), Kotias (10 kya), Ranchot88 (10 kya), and Stuttgart (8 kya), SI Appendix, Fig. S6] (8, 19). Our finding that there is no ongoing decline in Neandertal ancestry in Europeans suggests that Neandertal ancestry in Europe has not been diluted in a significant way by gene flow from Basal Eurasians. Specifically, we find no difference in Neandertal ancestry in European individuals with and without Basal Eurasian ancestry (direct f4-ratio mean 2.31% vs. 2.38%, respectively; P = 0.36). However, given the small number of relevant samples we also cannot exclude that there could be up to 13% less Neandertal ancestry in individuals with Basal Eurasian ancestry, or as much as 6% more Neandertal ancestry in individuals without Basal Eurasian ancestry (95% CI).In contrast, we do find that present-day Near Easterners carry significantly less Neandertal ancestry than Europeans (direct f4-ratio mean 2.03% vs. 2.33%; P = 0.001; SI Appendix, Fig. S7A). Furthermore, present-day populations in the Near East show even stronger signals of admixture with a deeply divergent modern human lineage than observed in the rest of West Eurasians (SI Appendix, Fig. S7B), suggesting that they carry additional ancestry components that are not present in Europe and that could potentially contribute to lower Neandertal ancestry in the Near East. We note, however, that a simple model of admixture from Africa into Near East would be expected to produce a similar f4 statistics difference between Near East and the rest of West Eurasia and could also explain lower values of Neandertal ancestry in this population. Long-Term Dynamics of Selection Against Introgressed DNA. Our observation that Neandertal ancestry levels did not significantly decrease from ∼45,000 y ago until today is seemingly at odds with the hypothesis that lower effective population sizes in Neandertals led to an accumulation of deleterious alleles, which were then subjected to negative selection in modern humans (3, 8⇓–10). To investigate the expected long-term dynamics of selection against Neandertal introgression under this hypothesis, we simulated a model of the human genome with empirical distributions of functional regions and selection coefficients, extending a strategy previously applied by Harris and Nielsen (6). We simulated modern human and Neandertal demography, including a low long-term effective population size (Ne) in Neandertals (Neandertal Ne = 1,000 vs. modern human Ne = 10,000) and 10% introgression at 55 kya (2,200 generations ago, assuming generation time of 25 y). To track the changes in Neandertal ancestry following introgression, we placed fixed Neandertal–human differences as neutral markers, both outside regions that accumulated deleterious mutations (to study the effect of negative selection on linked genome-wide neutral Neandertal variation) as well as within regions directly under selection (to track the effect of negative selection itself) (Fig. 3A).  Fig. 3. Simulations of selection against Neandertal ancestry. (A) Deleterious mutations (lightning bolts) accumulate in realistically distributed exonic sequence in modern humans and Neandertals. These regions accumulate additive, deleterious mutations, using a mutation rate of 10−8 per base pair per generation. To track the dynamics of Neandertal ancestry over time, neutral Neandertal markers are placed within (blue dots) and between (red dots) exons on all Neandertal chromosomes before introgression. (B) Simulated Neandertal ancestry proportions across 55 ky, in exonic and nonexonic sequence, averaged over 20 simulation replicates. Empirical observations from Fig. 1A are shown for comparison. Initial introgression levels were simulated at 10%. (C) Depletion of simulated Neandertal ancestry at neutral markers over time as a function of distance to regions under selection. Markers in bin 0 are those falling within exons; bins 1–5 represent quintiles of distance to the nearest exon. (D) Changes in frequencies of neutral Neandertal markers and deleterious Neandertal mutations over time, starting from generation 200. Each line shows average allele frequency changes over one simulation replicate. Black lines show smooth fits of these averages over 20 replicates. Similar to Harris and Nielsen (6), we observed abrupt removal of Neandertal alleles from the modern human population during the first ∼10 generations after introgression, followed by quick stabilization of Neandertal ancestry levels (Fig. 3B). Compared with empirical estimates of Neandertal ancestry, we find a better fit between these simulations and the direct f4-ratio estimate than with the indirect f4-ratio estimate, suggesting that our direct Neandertal ancestry estimates are consistent with theoretical expectations of genome-wide selection against introgression (Fig. 3B). Specifically, simulations show −0.004% change in Neandertal ancestry over 45 ky; in the empirical data this slope is not rejected using the direct f4-ratio (P = 0.29), but is significantly different from the indirect f4-ratio (P < 0.001). Because many factors can potentially influence the efficacy of negative selection, and no model fully captures all of these, we next sought to determine whether there is a combination of model parameters that could potentially lead to long-term continuous removal of Neandertal ancestry over time. Surprisingly, we failed to find a model which would produce a significant decline over time, although we tried by: (i) decreasing the long-term Neandertal Ne before introgression (making purifying selection in Neandertals even less efficient), (ii) increasing the Ne of modern humans after introgression (i.e., increasing the efficacy of selection against introgressed alleles), (iii) artificially increasing the deleteriousness of Neandertal variants after introgression (approximating a “hybrid incompatibility” scenario), (iv) simulating mixtures of dominance coefficients, or by (v) increasing the total amount of functional sequence (thereby increasing the number of accumulated deleterious variants in Neandertals and modern humans) (SI Appendix, Figs. S9–S13). Varying these factors primarily affected the magnitude of the initial removal of introgressed DNA by increasing the number of perfectly linked deleterious mutations in early Neandertal–modern human offspring (decreasing their fitness compared with individuals with less Neandertal ancestry), which in turn influenced the final level of Neandertal ancestry in the population (SI Appendix, Figs. S9–S13). The depletion of Neandertal ancestry around functional genomic elements in modern human genomes has also been taken as evidence for selection against Neandertal introgressed DNA (3, 8). We next examined the genomic distribution of Neandertal markers at different time points in our simulations to determine whether our models can recapitulate these signals. In agreement with empirical results in present-day humans (3), we found a strong negative correlation between the proportion of Neandertal introgression surviving at a locus and distance to the nearest region under selection (Fig. 3C). Furthermore, we found that the strength of this correlation increases over time, with the bulk of these changes occurring between 10 and 400 generations postadmixture [mean Pearson’s correlation coefficient ρ = 0.07, 0.79, 0.96 at generations 10, 400, and 2,200, respectively (SI Appendix, Fig. S15)]. We note that this time period predates all existing ancient modern human sequences, frustrating any current comparison with empirical data. However, despite no apparent change in genome-wide Neandertal ancestry proportion over time, we observe a smaller though still significant decrease in linked Neandertal ancestry during the time period for which modern human sequences exist (∼400–2,200 generations post-admixture) (Fig. 3 C and B). Indeed, by looking at the average per-generation changes in frequencies of simulated Neandertal mutations (that is, derivatives of allele frequencies in each generation), we observe the impact of negative selection on linked neutral Neandertal markers until at least ∼700 generations post admixture (Fig. 3D) and find that it closely follows the pattern of introgressed deleterious mutations (Fig. 3D). After this period of gradual removal, selection against linked neutral variation slows down significantly as genome-wide Neandertal ancestry becomes largely unlinked from regions that are under negative selection (Fig. 3D). In contrast, the selected variants themselves are still removed, although at increasingly slower rates (Fig. 3D). Due to this slow rate, and the small contribution these alleles make to genome-wide Neandertal ancestry, their continued removal has little impact on the slope of Neandertal ancestry over time. |
|
|
|
Post by Admin on Apr 23, 2019 17:48:17 GMT
Neandertal DNA Is Depleted in Regulatory and Conserved Noncoding Sequence. We next sought to leverage the direct f4-ratio in analyses of selection against introgression in functional genomic regions. Although previous studies have identified a depletion of Neandertal DNA in genomic regions with a high degree of evolutionary conservation, these studies have relied on maps of introgressed haplotypes (3, 29). Such maps may lack power to detect introgressed Neandertal DNA in highly conserved regions, as these regions may contain fewer informative sites carrying Neandertal–modern human differences. Furthermore, previous studies of negative selection against introgressed Neandertal DNA divided the genome into bins based on measures of evolutionary conservation, such as B values (30), which are not easily interpreted in terms of functional significance. To determine whether particular functional classes of genomic sites are differently affected by Neandertal introgression, we partitioned the human genome by functional annotation obtained from Ensembl v91 (31), and by primate conserved regions inferred using phastCons (32). For each annotation category, we estimated the Neandertal ancestry proportion in non-African Simons Genome Diversity Project (SGDP) individuals (excluding Oceanians) using the direct f4-ratio (Fig. 4).  Fig. 4. Neandertal ancestry estimates by genomic region. (Top) Direct f4-ratio estimates of Neandertal ancestry in all non-African SGDP individuals except Oceanians (known to carry Denisovan ancestry in addition to Neandertal ancestry) (25), with SNPs partitioned by functional annotation (Ensembl) or conservation (phastCons); “gap” combines intronic and intergenic sequence (dashed black line). Many annotation categories overlap other categories (SI Appendix, Table S1)—the largest is the 62% of protein-coding sequence which overlaps phastCons conserved elements (translucent orange). To minimize the noise in Neandertal ancestry estimates for small subsets of the genome, we calculated the direct f4-ratio using all SGDP Africans, except those that carry a high proportion of Neandertal alleles (Mozabite, Saharawi, Ju/’hoan North, Khomani San and Somali in SI Appendix, Fig. S4). Gray dashed line shows mean Neandertal ancestry in conserved phastCons regions. (Bottom) Idealized representation of genomic regions. In seeming contrast with previous studies (3, 8), we observed no significant depletion of Neandertal ancestry in CDS compared with intronic and intergenic regions (referred to as “gap” regions below) (average direct f4-ratio ∼1.94% in both; Fig. 4). However, we did identify a striking depletion of Neandertal ancestry in both promoters and phastCons conserved regions (1.15% and 0.95%), with both containing significantly less Neandertal ancestry than gap regions (P = 0.004 and P < 0.0001, estimated via resampling as described in SI Appendix, section S1). We note that 62% of CDS overlaps with phastCons regions (21% of phastCons conserved tracks overlap CDS); indeed, conserved CDS has a lower Neandertal ancestry estimate (1.25%) than overall CDS, although not as low as all phastCons regions (Fig. 4). These results suggest that previously observed depletions in conserved and genic regions may not have been driven primarily by protein-coding differences between Neandertals and modern humans, as was previously assumed, but rather by differences in promoters and other noncoding conserved sequence. This hypothesis is supported by several recent studies of the effects of introgressed Neandertal sequences, including those with signatures of adaptive introgression, which found that surviving functional introgressed haplotypes have their major influence on gene expression regulation (33⇓⇓⇓–37). We note that the lack of a depletion in CDS does not fit the observations from our simulations (Fig. 3C). Assuming additivity, and a distribution of fitness effects (DFEs) derived from the frequency spectra of mutations altering coding sequence (38), these simulations predict a reduction of 5–17% Neandertal ancestry versus nonselected regions, depending on distance from selected regions (Fig. 3C). In addition, the reduction in simulations is much smaller than the empirical depletions of promoter and phastCons regions (40% and 51%, respectively). Together, these demonstrate that the actions of selection against Neandertal sequence are not fully captured by the models presented here. Although it is beyond the scope of this work, it may be possible to leverage distributions of Neandertal ancestry in studying the action of selection in noncoding sequence. Challenges associated with such work include the uncertainty of the DFE of mutations affecting noncoding sequence, and their dominance coefficients, potential epistatic effects of regulatory mutations, as well as the fact that a single deleterious mutation can affect a region falling into multiple functional categories at once (SI Appendix, Table S1). Conclusions Our reevaluation of Neandertal ancestry in modern human genomes indicates that overall levels of Neandertal ancestry in Europe have not significantly decreased over the past 45,000 y, and that previous observations of continuous Neandertal ancestry decline were likely an artifact of unaccounted-for gene flow increasing allele sharing between West Eurasian and African populations. Nevertheless, we do find evidence of selection against Neandertal DNA in the genome-wide distribution of Neandertal ancestry, with such ancestry depleted in promoter and other noncoding conserved DNA more strongly than in protein-coding sequence, raising the possibility that Neandertals may have differed more from modern humans in their regulatory variants than in their protein-coding sequences, and that regulatory variation may provide a richer template for selection to act upon. Furthermore, simulations suggest that negative selection against introgression is expected to have the strongest impact on genome-wide Neandertal ancestry during the first few hundred generations, before the time frame for which ancient samples are currently available. The genomes of early modern humans living 55–50 kya, although difficult to obtain, may shed additional light on the process of selection against Neandertal DNA, as well as on early out-of-Africa demography. Our findings can be extrapolated to other cases where one species or population contributes a fraction of ancestry to another species or population, a frequent occurrence in nature (5, 29, 39⇓–41). Even in cases where the introgressing population carries a high burden of deleterious mutations, negative selection is not expected to result in an extended decrease in the overall genome-wide ancestry contributed by that population. Therefore, any long-term shifts in overall ancestry proportions over time are likely to be the result of forces other than negative selection, for example admixture with one or more other populations. PNAS January 29, 2019 116 (5) 1639-1644; published ahead of print January 29, 2019 |
|

