|
|
Post by Admin on Apr 6, 2019 22:59:34 GMT
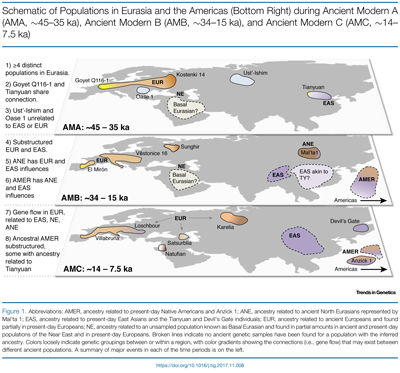 Eurasia ∼45–35ka shows the presence of at least four distinct populations: early Asians and Europeans, as well as populations with ancestry found hardly or not at all in present-day populations. Europeans from around 34–15ka show high internal population structure. Approximately 14–7.5ka, populations across Eurasia shared genetic similarities, suggesting greater interactions between geographically distant populations. Ancient modern human genomes support at least two Neanderthal admixture events, one ∼60–50ka in early ancestors of non-African populations and a second >37ka related to the Oase 1 individual. In the Caucasus, ~13–10 000-year-old individuals (i.e., Satsurblia and Kotias) show a close relation-ship to ancient individuals in West Eurasia, but they also possess the Basal Eurasian ancestry observed in populations from Europe and the Near East [13,27,28] (Figure 2B). Basal Eurasian ancestry is highest in the Near East, with estimates as high as 66% in Epipaleolithic Natufianindividuals from the Levant ~12–9.8 ka, and 44% in a Mesolithic individual from Iran from ~9.1to 8.6 ka (i.e., Hotu) [28]. Further sampling will help to determine whether the gene flow between populations in the AMC began during this time period or extends back into the AMB.  Before Australian Aborigines and New Guineans and South Indians and Native Americans and other indigenous hunter-gatherers split, they split from Basal Eurasians. Early European farmers had ∼44% ancestry from a ‘Basal Eurasian’ population that split before the diversification of other non-African lineages.  The area of the Near East where Basal Eurasians resided was the Arabian Peninsula, from where Haplogroup E spread to the Near East and North Africa. There was no contact with Neanderthals in the Arabian Peninsula as it was outside the Neanderthal range, which explains why Near Easterners carry significantly less Neanderthal ancestry than Europeans. There was a sharp genetic transition between the hunter-gatherers and the farmers, reflecting a major movement of new people into Europe from the Near East. 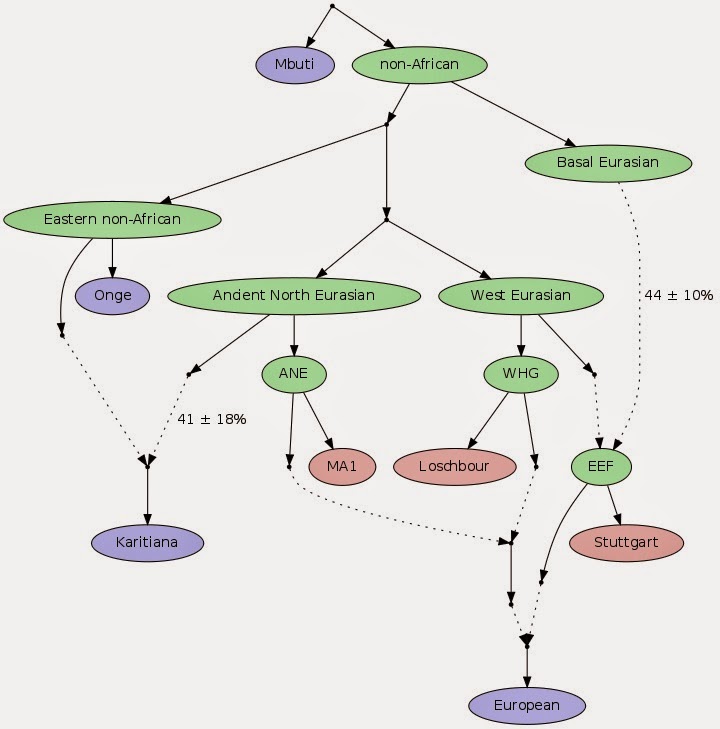 Haplogroup E is the Basal Eurasian haplogroup, based on its frequencies in the Near East (10%) and Northern Europe (1%). The Bedouin are the descendants of the earliest split from Basal Eurasian populations (Rodriguez-Flores et al. 2016) with the high frequency of Haplogroup E-M123 (24.3 %). The Natufians (50% Basal Eurasian) carried E-Z830, an ancestral clade of E-M123. A Saudi Arabian study found that Y chromosome DE-YAP* originated in the Arabian Peninsula, which served as an incubator for the early diversification of non-African haplogroups (Abu-Amero et al. 2009). It's plausible that DE-YAP* evolved in the Arabian Peninsula around 65,000 years ago and Haplogroup D subsequently headed for Asia and Oceania from the Arabian Peninsula after splitting with E along with the three main Eurasian mtDNA haplogroups M, R, and N.  East Asians can be explained by dilution of Neandertal ancestry in Europeans due to admixture with a hypothetical Basal Eurasian population that carried little to no Neandertal ancestry (19, 28). Previous studies have found Basal Eurasian ancestry in all modern and some ancient Europeans [in this study, four ancient individuals show evidence of Basal Eurasian ancestry: Satsurblia (15 kya), Kotias (10 kya), Ranchot88 (10 kya), and Stuttgart (8 kya), SI Appendix, Fig. S6] (8, 19). Our finding that there is no ongoing decline in Neandertal ancestry in Europeans suggests that Neandertal ancestry in Europe has not been diluted in a significant way by gene flow from Basal Eurasians. In contrast, we do find that present-day Near Easterners carry significantly less Neandertal ancestry than Europeans (direct f4-ratio mean 2.03% vs. 2.33%; P = 0.001; SI Appendix, Fig. S7A).  Furthermore, present-day populations in the Near East show even stronger signals of admixture with a deeply divergent modern human lineage than observed in the rest of West Eurasians (SI Appendix, Fig. S7B), suggesting that they carry additional ancestry components that are not present in Europe and that could potentially contribute to lower Neandertal ancestry in the Near East. We note, however, that a simple model of admixture from Africa into Near East would be expected to produce a similar f4 statistics difference between Near East and the rest of West Eurasia and could also explain lower values of Neandertal ancestry in this population (Petr et al. 2019).  |
|
|
|
Post by Admin on Apr 7, 2019 17:53:13 GMT
Near Eastern migrants played a major role in the introduction of agriculture to Europe, as ancient DNA indicates that early European farmers were distinct from European hunter-gatherers4,5 and close to present-day Near Easterners4,6. However, modelling present-day Europeans as a mixture of these two ancestral populations4 does not account for the fact that they are also admixed with a population related to Native Americans7,8. To clarify the prehistory of Europe, we sequenced nine ancient genomes (Fig. 1A; Extended Data Fig. 1): “Stuttgart” (19-fold coverage), a ~7,000 year old skeleton found in Germany in the context of artifacts from the first widespread farming culture of central Europe, the Linearbandkeramik; “Loschbour” (22-fold), an ~8,000 year old skeleton from the Loschbour rock shelter in Luxembourg, discovered in the context of hunter-gatherer artifacts (SI1; SI2); and seven ~8,000 year old samples (0.01–2.4-fold) from a hunter-gatherer burial in Motala, Sweden (the highest coverage individual was “Motala12”). Figure 1: Map of west Eurasian populations. Sequence reads from all samples revealed >20% C→T and G→A deamination-derived mismatches at the ends of the molecules that are characteristic of ancient DNA9,10 (SI3). We estimate nuclear contamination rates to be 0.3% for Stuttgart and 0.4% for Loschbour (SI3), and mitochondrial (mtDNA) contamination rates to be 0.3% for Stuttgart, 0.4% for Loschbour, and 0.01–5% for the Motala individuals (SI3). Stuttgart has mtDNA haplogroup T2, typical of Neolithic Europeans11, and Loschbour and all Motala individuals have the U5 or U2 haplogroups, typical of hunter-gatherers5,9 (SI4). Stuttgart is female, while Loschbour and five Motala individuals are male (SI5) and belong to Y-chromosome haplogroup I, suggesting that this was common in pre-agricultural Europeans (SI5). We carried out large-scale sequencing of libraries prepared with uracil DNA glycosylase (UDG), which removes deaminated cytosines, thus reducing errors arising from ancient DNA damage (SI3). The ancient individuals had indistinguishable levels of Neanderthal ancestry when compared to each other (~2%) and to present-day Eurasians (SI6). The heterozygosity of Stuttgart (0.00074) is at the high end of present-day Europeans, while that of Loschbour (0.00048) is lower than in any present humans (SI2), reflecting a strong bottleneck in Loschbour’s ancestors as the genetic data show that he was not recently inbred (Extended Data Fig. 2). High copy numbers for the salivary amylase gene (AMY1) have been associated with a high starch diet12; our data are consistent with this finding in that the ancient hunter gatherers La Braña (from Iberia)2, Motala12, and Loschbour had 5, 6 and 13 copies respectively, whereas the Stuttgart farmer had 16 (SI7). Both Loschbour and Stuttgart had dark hair (>99% probability); and Loschbour, like La Braña and Motala12, likely had blue or intermediate-colored eyes (>75%) while Stuttgart likely had brown eyes (>99%) (SI8). Neither Loschbour nor La Braña carries the skin-lightening allele in SLC24A5 that is homozygous in Stuttgart and nearly fixed in Europeans today2, but Motala12 carries at least one copy of the derived allele, showing that this allele was present in Europe prior to the advent of agriculture.  Figure 2: Principal Component Analysis. We compared the ancient genomes to 2,345 present-day humans from 203 populations genotyped at 594,924 autosomal single nucleotide polymorphisms (SNPs) with the Human Origins array8 (SI9) (Extended Data Table 1). We used ADMIXTURE13 to identify 59 “West Eurasian” populations that cluster with Europe and the Near East (SI9 and Extended Data Fig. 3). Principal component analysis (PCA)14 (SI10) (Fig. 1B) indicates a discontinuity between the Near East and Europe, with each showing north-south clines bridged only by a few populations of mainly Mediterranean origin. We projected15 the newly sequenced and previously published1–4 ancient genomes onto the first two principal components (PCs) (Fig. 1B). Upper Paleolithic hunter-gatherers3 from Siberia like the MA1 (Mal’ta) individual project at the northern end of the PCA, suggesting an “Ancient North Eurasian” meta-population (ANE). European hunter-gatherers from Spain2, Luxembourg, and Sweden4 fall beyond present-day Europeans in the direction of European differentiation from the Near East, and form a “West European Hunter-Gatherer” (WHG) cluster including Loschbour and La Braña2, and a “Scandinavian Hunter-Gatherer” (SHG) cluster including the Motala individuals and ~5,000 year old hunter-gatherers from the Pitted Ware Culture4. An “Early European Farmer” (EEF) cluster includes Stuttgart, the ~5,300 year old Tyrolean Iceman1 and a ~5,000 year old Swedish farmer4. Patterns observed in PCA may be affected by sample composition (SI10) and their interpretation in terms of admixture events is not straightforward, so we rely on formal analysis of f-statistics8 to document mixture of at least three source populations in the ancestry of present Europeans. We began by computing all possible statistics of the form f3(Test; Ref1, Ref2) (SI11), which if significantly negative show unambiguously8 that Test is admixed between populations anciently related to Ref1 and Ref2 (we choose Ref1 and Ref2 from 5 ancient and 192 present populations). The lowest f3-statistics for Europeans are negative (93% are >4 standard errors below 0), with most showing strong support for at least one ancient individual being one of the references (SI11). Europeans almost always have their lowest f3 with either (EEF, ANE) or (WHG, Near East) (SI11, Table 1, Extended Data Table 1), which would not be expected if there were just two ancient sources of ancestry (in which case the best references for all Europeans would be similar). The lowest f3-statistic for Near Easterners always takes Stuttgart as one of the reference populations, consistent with a Near Eastern origin for Stuttgart’s ancestors (Table 1). We also computed the statistic f4(Test, Stuttgart; MA1, Chimp), which measures whether MA1 shares more alleles with a Test population or with Stuttgart. This statistic is significantly positive (Extended Data Fig. 4, Extended Data Table 1) if Test is nearly any present-day West Eurasian population, showing that MA1-related ancestry has increased since the time of early farmers like Stuttgart (the analogous statistic using Native Americans instead of MA1 is correlated but smaller in magnitude (Extended Data Fig. 5), indicating that MA1 is a better surrogate than the Native Americans who were first used to document ANE ancestry in Europe7,8). The analogous statistic f4(Test, Stuttgart; Loschbour, Chimp) is nearly always positive in Europeans and negative in Near Easterners, indicating that Europeans have more ancestry from populations related to Loschbour than do Near Easterners (Extended Data Fig. 4, Extended Data Table 1). Extended Data Table 2 documents the robustness of key f4-statistics by recomputing them using transversion polymorphisms not affected by ancient DNA damage, and also using whole-genome sequencing data not affected by SNP ascertainment bias. Extended Data Fig. 6 shows the geographic gradients in the degree of allele sharing of present-day West Eurasians (as measured by f4-statistics) with Stuttgart (EEF), Loschbour (WHG) and MA1 (ANE). To determine the minimum number of source populations needed to explain the data for many European populations taken together, we studied the matrix of all possible statistics of the form f4(Testbase, Testi; Obase, Oj) (Supplementary Information section 12). Testbase is a reference European population, Testi is the set of all other European Test populations, Obase is a reference outgroup, and Oj is the set of other outgroups (ancient DNA samples, Onge, Karitiana, and Mbuti). The rank of the (i, j) matrix reflects the minimum number of sources that contributed to the Test populations16,17. For a pool of individuals from 23 Test populations representing most present-day European groups, this analysis rejects descent from just two sources (P < 10−12 by a Hotelling t-test17). However, three source populations are consistent with the data after excluding the Spanish who have evidence for African admixture18,19,20 (P = 0.019, not significant after multiple-hypothesis correction), consistent with the results from ADMIXTURE (Supplementary Information section 9), PCA (Fig. 2 and Supplementary Information section 10) and f statistics (Extended Data Table 1, Extended Data Fig. 6, Supplementary Information sections 11 and 12). We caution that the finding of three sources could be consistent with a larger number of mixture events. Moreover, the source populations may themselves have been mixed. Indeed, the positive f4(Stuttgart, Test; Loschbour, Chimp) statistics obtained when Test is Near Eastern (Extended Data Table 1) imply that the EEF had some WHG-related ancestry, which was greater than 0% and as high as 45% (Supplementary Information section 13). |
|
|
|
Post by Admin on Apr 7, 2019 20:34:00 GMT
We used the ADMIXTUREGRAPH software8,15 to fit a model (a tree structure augmented by admixture events) to the data, exploring models relating the three ancient populations (Stuttgart, Loschbour, and MA1) to two eastern non-Africans (Onge and Karitiana) and sub-Saharan Africans (Mbuti). We found no models that fit the data with 0 or 1 admixture events, but did find a model that fit with 2 admixture events (SI14). The successful model (Fig. 2A) confirms the existence of MA1-related admixture in Native Americans3, but includes the novel inference that Stuttgart is partially (44 ± 10%) derived from a lineage that split prior to the separation of eastern non-Africans from the common ancestor of WHG and ANE. The existence of such “Basal Eurasian” admixture into Stuttgart provides a simple explanation for our finding that diverse eastern non-African populations share significantly more alleles with ancient European and Upper Paleolithic Siberian hunter-gatherers than with Stuttgart (that is, f4(Eastern non-African, Chimp; Hunter-gatherer, Stuttgart) is significantly positive), but that hunter-gatherers appear to be equally related to most eastern groups (SI14). We verified the robustness of the model by reanalyzing the data using the unsupervised MixMapper7 (SI15) and TreeMix21 software (SI16), which both identified the same admixture events. The ANE/WHG split must have occurred >24,000 years ago (as it must predate the age of MA13), and the WHG/Eastern non-African split must have occurred >40,000 years ago (as it must predate the Tianyuan22 individual from China which clusters with Asians to the exclusion of Europeans). The Basal Eurasian split must be even older, and might be related to early settlement of the Levant23 or Arabia24,25 prior to the diversification of most Eurasians, or more recent gene flow from Africa26. However, the Basal Eurasian population shares much of the genetic drift common to non-African populations after their separation from Africans, and thus does not appear to represent gene flow between sub-Saharan Africans and the ancestors of non-Africans after the out-of-Africa bottleneck (SI14).  Figure 3: Modelling the relationship of European to non-European populations. Fitting present-day Europeans into the model, we find that few populations can be fit as 2-way mixtures, but nearly all are compatible with 3-way mixtures of ANE/EEF/WHG (SI14). The mixture proportions from the fitted model (Fig. 2B; Extended Data Table 3) are encouragingly consistent with those obtained from a separate method that relates European populations to diverse outgroups using f4-statistics, assuming only that MA1 is an unmixed descendent of ANE, Loschbour of WHG, and Stuttgart of EEF (SI17). We infer that EEF ancestry in Europe today ranges from ~30% in the Baltic region to ~90% in the Mediterranean, consistent with patterns of identity-by-descent (IBD) sharing27,28 (SI18) and shared haplotype analysis (chromosome painting)29 (SI19) in which Loschbour shares more segments with northern Europeans and Stuttgart with southern Europeans. Southern Europeans inherited their European hunter-gatherer ancestry mostly via EEF ancestors (Extended Data Fig. 6), while Northern Europeans acquired up to 50% of WHG ancestry above and beyond the WHG-related ancestry which they received through their EEF ancestors. Europeans have a larger proportion of WHG than ANE ancestry in general. By contrast, in the Near East there is no detectable WHG ancestry, but up to ~29% ANE in the Caucasus (SI14). A striking feature of these findings is that ANE ancestry is inferred to be present in nearly all Europeans today (with a maximum of ~20%), but was absent in both farmers and hunter-gatherers from central/western Europe during the Neolithic transition. At the same time, we infer that ANE ancestry was not completely absent from the larger European region at that time: we find that it was present in ~8,000 years old Scandinavian hunter-gatherers, since MA1 shares more alleles with Motala12 (SHG) than with Loschbour, and Motala12 fits as a mixture of 81% WHG and 19% ANE (SI14). Two sets of European populations are poor fits for the model. Sicilians, Maltese, and Ashkenazi Jews have EEF estimates of >100% consistent with their having more Near Eastern ancestry than can be explained via EEF admixture (SI17). They also cannot be jointly fit with other Europeans (SI14), and they fall in the gap between European and Near Easterners (Fig. 1B). Finns, Mordovians and Russians (from the northwest of Russia) also do not fit (SI14; Extended Data Table 3) due to East Eurasian gene flow into the ancestors of these northeastern European populations. These populations (and Chuvash and Saami) are more related to East Asians than can be explained by ANE admixture (Extended Data Fig. 7), likely reflecting a separate stream of Siberian gene flow into northeastern Europe (SI14).  Figure 4: Estimates of mixture proportions in present-day Europeans. Fitting present-day Europeans into the model, we find that few populations can befit as two-way mixtures, but nearly all are compatible with three-way mixtures of ANE–EEF–WHG (Supplementary Information section 14). The mixture proportions from the fitted model (Fig. 4 and Extended Data Table 3) are encouragingly consistent with those obtained from a separate method that relates European populations to diverse outgroups using f4 statistics, assuming only that MA1 is an unmixed descendent of ANE, Loschbour of WHG, and Stuttgart of EEF (Supplementary Information section 17). We infer that EEF ancestry in Europe today ranges from ,30% in the Baltic region to ,90% in the Mediterranean, consistent with patterns of identity-by-descent (IBD) sharing27,28 (Supplementary Information section 18) and shared haplotype analysis (chromosome painting)29 (Supplementary Information section 19) in which Loschbour shares more segments with northern Europeans and Stuttgart with southern Europeans. Southern Europeans inherited their European hunter-gatherer ancestry mostly via EEF ancestors (Extended Data Fig. 6), whereas northern Europeans acquired up to 50% of WHG ancestry above and beyond what they received through their EEF ancestors. Europeans have a larger proportion of WHG than ANE ancestry in general. By contrast, in the Near East there is no detectable WHG ancestry, but up to ,29% ANE in the Caucasus (Supplementary Information section 14). A striking feature of these findings is that ANE ancestry is inferred to be present in nearly all Europeans today (with a maximum of 20%), but was absent in both farmers and hunter-gatherers from central and western Europe during the Neolithic transition. However, ANE ancestry was not completely absent from the larger European region at that time: we find that it was present in ,8,000-years-old Scandinavian hunter-gatherers, as MA1 shares more alleles with Motala12 (SHG) than with Loschbour, and Motala12 fits as a mixture of 81% WHG and 19% ANE (Supplementary Information section 14). Two sets of European populations are poor fits for the model. Sicilians, Maltese, and Ashkenazi Jews have EEF estimates of .100%, consistent with their having more Near Eastern ancestry than can be explained via EEF admixture (Supplementary Information section 17). They also cannot be jointly fit with other Europeans (Supplementary Information section 14), and they fall in the gap between European and Near Easterners in PCA (Fig. 2). Finns, Mordovians and Russians (from the northwest of Russia) also do not fit (Supplementary Information section 14; Extended Data Table 3) due to East Eurasian gene flow into the ancestors of these north-eastern European populations. These populations (and Chuvash and Saami) are more related to east Asians than can be explained by ANE admixture (Extended Data Fig. 7), probably reflecting a separate stream of Siberian gene flow into north-eastern Europe (Supplementary Information section 14). Several questions will be important to address in future ancient DNA work. Where and when did the Near Eastern farmers admix with European hunter-gatherers to produce the EEF? How did the ancestors of present-day Europeans first acquire their ANE ancestry? Discontinuity in central Europe during the late Neolithic (~4,500 years ago) associated with the appearance of mtDNA types absent in earlier farmers and hunter-gatherers30 raises the possibility that ANE ancestry may have also appeared at this time. Finally, it is important to study ancient genome sequences from the Near East to provide insights into the history of the Basal Eurasians. Nature. 2014 Sep 18; 513(7518): 409–413. |
|
|
|
Post by Admin on Apr 8, 2019 17:53:18 GMT
An open question in the history of human migration is the identity of the earliest Eurasian populations that have left contemporary descendants. The Arabian Peninsula was the initial site of the out-of-Africa migrations that occurred between 125,000 and 60,000 yr ago, leading to the hypothesis that the first Eurasian populations were established on the Peninsula and that contemporary indigenous Arabs are direct descendants of these ancient peoples. To assess this hypothesis, we sequenced the entire genomes of 104 unrelated natives of the Arabian Peninsula at high coverage, including 56 of indigenous Arab ancestry. The indigenous Arab genomes defined a cluster distinct from other ancestral groups, and these genomes showed clear hallmarks of an ancient out-of-Africa bottleneck. Similar to other Middle Eastern populations, the indigenous Arabs had higher levels of Neanderthal admixture compared to Africans but had lower levels than Europeans and Asians. These levels of Neanderthal admixture are consistent with an early divergence of Arab ancestors after the out-of-Africa bottleneck but before the major Neanderthal admixture events in Europe and other regions of Eurasia. When compared to worldwide populations sampled in the 1000 Genomes Project, although the indigenous Arabs had a signal of admixture with Europeans, they clustered in a basal, outgroup position to all 1000 Genomes non-Africans when considering pairwise similarity across the entire genome. These results place indigenous Arabs as the most distant relatives of all other contemporary non-Africans and identify these people as direct descendants of the first Eurasian populations established by the out-of-Africa migrations.  All humans can trace their ancestry back to Africa (Cann et al. 1987), where the ancestors of anatomically modern humans first diverged from primates (Patterson et al. 2006), and then from archaic humans (Prüfer et al. 2014). Humans began leaving Africa through a number of coastal routes, where estimates suggest these “out-of-Africa” migrations reached the Arabian Peninsula as early as 125,000 yr ago (Armitage et al. 2011) and as late as 60,000 yr ago (Henn et al. 2012). After entering the Arabian Peninsula, human ancestors entered South Asia and spread to Australia (Rasmussen et al. 2011), Europe, and eventually, the Americas. The individuals in these migrations were the most direct ancestors of ancient non-African peoples, and they established the contemporary non-African populations recognized today (Cavalli-Sforza and Feldman 2003). The relationship between contemporary Arab populations and these ancient human migrations is an open question (Lazaridis et al. 2014; Shriner et al. 2014). Given that the Arabian Peninsula was an initial site of egress from Africa, one hypothesis is that the original out-of-Africa migrations established ancient populations on the peninsula that were direct ancestors of contemporary Arab populations (Lazaridis et al. 2014). These people would therefore be direct descendants of the earliest split in the lineages that established Eurasian and other contemporary non-African populations (Armitage et al. 2011; Rasmussen et al. 2011; Henn et al. 2012; Lazaridis et al. 2014; Shriner et al. 2014). If this hypothesis is correct, we would expect that there are contemporary, indigenous Arabs who are the most distant relatives of other Eurasians. To assess this hypothesis, we carried out deep-coverage genome sequencing of 104 unrelated natives of the Arabian Peninsula who are citizens of the nation of Qatar (Supplemental Fig. 1), including 56 of indigenous Bedouin ancestry who are the best representatives of autochthonous Arabs, and compared these genomes to contemporary genomes of Africa, Asia, Europe, and the Americas (The 1000 Genomes Project Consortium 2012; Lazaridis et al. 2014).  Figure 1. Principal component analysis (PCA) (Price et al. 2006) of the 104 Qatari genomes (circle), 1000 Genomes (triangle), and Human Origins (square) study samples. Shown are individuals plotted on principal components PC1 and PC2, with genomes color-coded by study and population, with the Q0 (Subpopulation Unassigned) in gray, Q1 (Bedouin) in red, Q2 (Persian-South Asian) in azure, and Q3 (African) in black. An analysis of inbreeding for these remaining individuals showed the Q1 (Bedouin) to have a more positive inbreeding coefficient than most of the non-admixed 1000 Genomes (The 1000 Genomes Project Consortium 2012) populations (Supplemental Table IV; Supplemental Fig. 4), consistent with the known inbreeding of this group (Hunter-Zinck et al. 2010; Omberg et al. 2012); although we also found the Q1 (Bedouin) to be less inbred than many small and/or isolated populations worldwide represented in the Human Origins samples (Lazaridis et al. 2014) (Supplemental Table V; Supplemental Table VI; Supplemental Fig. 4). The Q2 (Persian-South Asian) had a positive, but slightly lower, inbreeding coefficient than the Q1 (Bedouin). In contrast, the Q3 (African) had a non-negative coefficient that reflects known admixture with African populations (Hunter-Zinck et al. 2010; Omberg et al. 2012).  We confirmed the primary ancestry classifications of the 104 Qataris by principal component analysis (Price et al. 2006). We combined the 104 Qataris, the Human Origins populations (Lazaridis et al. 2014), and 1000 Genomes populations (The 1000 Genomes Project Consortium 2012) (excluding individuals already in Human Origins), and performed principal component analysis on a set of 197,714 linkage disequilibrium pruned autosome SNPs (Fig. 1A; Supplemental Fig. 5A). We also confirmed these clusterings just with the 104 Qataris and 1000 Genomes samples based on the same set of autosomal SNPs (Supplemental Fig. 5B). These analyses reproduced the population clustering observed previously (Hunter-Zinck et al. 2010; Omberg et al. 2012), with the Q1 (Bedouin) closest to Europeans, the Q2 (Persian-South Asian) between Q1 (Bedouin) and Asians, and the Q3 (African) closest to African populations. A plot of just the Middle Eastern populations on the principal components also showed clustering as expected, with the Q1 (Bedouin) clustering with previously sampled Bedouins and Arabs, Q2 (Persian-South Asians) with Iranians, and Q3 (African) outside of the Middle Eastern cluster (data not shown) (Fig. 1B). |
|
|
|
Post by Admin on Apr 9, 2019 18:27:49 GMT
Y Chromosome and mitochondrial DNA haplogroups We next analyzed the Y Chromosome (Chr Y) and mitochondrial DNA (mtDNA) to assess the degree to which the Q1 (Bedouin), Q2 (Persian-South Asian), or Q3 (African) Qatari ancestry groups represent distinct subpopulations (Fig. 2). The Chr Y haplogroups showed almost no overlap between the Q1 (Bedouin) Qataris and Q2 (Persian-South Asian) Qataris, in which an Analysis of Molecular Variance (AMOVA) was highly significant (P < 0.018) (Supplemental Table VII). The Arab haplogroup J1 was the dominant haplogroup in the Q1 (Bedouin) Qataris, but this haplogroup was not represented at all among the Q2 (Persian-South Asian) Qataris (Fig. 2A). This confirmed that these are genetically well-defined subpopulations that are relatively isolated from one another (Omberg et al. 2012). There was also a strong partitioning of the Chr Y haplogroups when considering the Q3 (African) Qataris, both when considering Q1 (Bedouin) versus Q3 (African) (AMOVA P < 1 × 10−5) and Q2 (Persian-South Asian) versus Q3 (African) (AMOVA P < 0.028). The Q3 (African) had largely African haplogroups, a result consistent with the known recent African admixture of this subpopulation (Omberg et al. 2012).  Figure 2. Y Chromosome (Chr Y) and mitochondrial DNA (mtDNA) haplogroup assignments. The Chr Y and mtDNA haplogroups were determined for Q1 (Bedouin), Q2 (Persian-South Asian), and Q3 (African). (A) Pie charts of the haplogroup frequencies for Chr Y. (B) Pie charts of the haplogroup frequencies for mtDNA. The mtDNA haplogroups were less partitioned among the Qataris, although they still showed significant partitioning between each pair of subpopulations (AMOVA Q1 versus Q2 P < 0.035, Q1 versus Q3 P < 1 × 10−5, Q2 versus Q3 P < 0.017) and among all three considered simultaneously (AMOVA P < 1 × 10−5) (Supplemental Table VII). The mtDNA haplogroups also included more worldwide geographic diversity overall, indicating a different male versus female pattern of intermarriage among these subpopulations (Sandridge et al. 2010). Together the Chr Y and mtDNA haplogroups indicate that the Q1 (Bedouin), Q2 (Persian-South Asian), and Q3 (African) ancestry groups represent genetic subpopulations that not only reflect known migration history (Hunter-Zinck et al. 2010; Omberg et al. 2012) but that also represent units defined by a patrilocal society with strong historical barriers to intermarriage (Esposito 2001; Cavalli-Sforza and Feldman 2003), in which gene flow has been dominated by female movement (i.e., admixture occurring through females marrying into the relatively isolated subpopulations), as well as female influxes from other geographic areas. X-linked and autosomal diversity To further analyze the relative male and female contributions to the genetics of the Qatari Q1 (Bedouin), Q2 (Persian-South Asian), and Q3 (African) subpopulations, we analyzed genome-wide ratios of X-linked and autosomal (X/A) diversity and X/A diversity ratios for genome intervals >0.18 cM from genes (Supplemental Table VIII; Supplemental Fig. 6). For both of these ratios, the Q1 (Bedouin) and Q2 (Persian-South Asian) were lower than for African populations but were higher than for Europeans and Asians. This points to a higher effective population size of females in the Q1 (Bedouin) and Q2 (Persian-South Asian), possibly a consequence of the out-of-Africa migrations, which were believed to be biased toward migration of males over females (Gottipati et al. 2011; Arbiza et al. 2014). The Q3 (African) Qataris had X/A diversity ratios that were higher, even when compared to African populations. This may be driven by a smaller male effective population size; a possible consequence of a polygamous culture and the ancestry of the Q3 (African) subpopulation that was a result of the historical slave trade into the region from Africa (Omberg et al. 2012). We also analyzed the relative ratios of X-linked and autosomal (X/A) diversity in nongenic regions of the female Q1 (Bedouin), Q2 (Persian-South Asian), and Q3 (African) genomes compared to females in African populations of the 1000 Genomes Project (Supplemental Table IX). The relative X/A ratios of both the Q1 (Bedouin) and Q2 (Persian-South Asian) to African populations were slightly higher than when comparing European to African populations (Gottipati et al. 2011; Arbiza et al. 2014). This could indicate a slightly less extreme set of bottleneck events encountered since the out-of-Africa migrations by the direct ancestors of the Q1 (Bedouin) and Q2 (Persian-South Asian) compared to the bottlenecks encountered by the direct ancestors of Europeans. The relative X/A diversity ratios of Q3 (African) to African populations were closer to one, consistent with the known African admixture of this subpopulation (Omberg et al. 2012).  Pairwise sequential Markov coalescent analysis We next analyzed the full complement of autosomal polymorphisms for signals of ancient bottlenecks by applying the pairwise sequential Markov coalescent (PSMC) (Fig. 3; Li and Durbin 2011). This analysis showed that the Q1 (Bedouin) and Q2 (Persian-South Asian) had clear hallmarks of a bottleneck event, with effective population size hitting a trough in the range of 100,000 to 30,000 yr ago with a minimum at ∼60,000 yr ago. This same pattern is observed for a European individual from the 1000 Genomes Project and is consistent with what has been observed in other non-African human genomes using the pairwise sequential Markov coalescent, as well as related methods (Gronau et al. 2011; Fu et al. 2014; Schiffels and Durbin 2014). These data, therefore, point to the ancestors of Q1 (Bedouin) and Q2 (Persian-South Asian) as having migrated out of Africa at the same time as the ancestors of other non-African populations (Henn et al. 2012). Although PSMC estimates in the more recent past tend to have larger confidence intervals (Li and Durbin 2011), the Q1 (Bedouin) do appear to have a lower population size than the Q2 (Persian-South Asian) in the region <30,000 yr ago, consistent with high levels of inbreeding in the Q1 (Bedouin) (Hunter-Zinck et al. 2010; Sandridge et al. 2010; Mezzavilla et al. 2015). For the Q3 (African), the median effective population size was more similar to an African individual from the 1000 Genomes Project in the range 100,000 to 30,000 yr ago, consistent with Sub-Saharan African ancestry that is relatively recent (Omberg et al. 2012).  Figure 3. Ancient bottlenecks in the 96 Q1 (Bedouin), Q2 (Persian-South Asian), or Q3 (African) Qatari genomes (56 Q1, 20 Q2, 20 Q3) determined by pairwise sequential Markov coalescent analysis (Li and Durbin 2011). Shown is the plot of the median effective population size (y-axis) across individuals in a subpopulation versus years in the past (log scale x-axis) for the samples in the three major Qatari subpopulations: Q1 (Bedouin) in red, Q2 (Persian-South Asian) in azure, Q3 (African) in black. A single individual of European ancestry (NA12879, violet) and a single individual of African ancestry (NA19239, orange) from the 1000 Genomes Project deep-coverage pilot (The 1000 Genomes Project Consortium 2010) are shown for comparison. Admixture analysis The signal of an ancient bottleneck in the Q1 (Bedouin) is not unexpected given previous analyses of genomic admixture that found <1% African ancestry in this subpopulation (Omberg et al. 2012) and studies of worldwide population structure, which have inferred that the Q1 (Bedouin) genomes have the greatest proportion of Arab genetic ancestry, even when compared to Bedouins from outside Qatar and to Arabs in surrounding countries, including Yemen and Saudi Arabia (Hodgson et al. 2014; Shriner et al. 2014). To confirm a similarly minute amount of African admixture for the Q1 (Bedouin) in our sample, we applied three methodologies: (1) an ADMIXTURE (Alexander et al. 2009) analysis of the genome-wide ancestry proportions in the 104 Qataris, the 1000 Genomes Project (The 1000 Genomes Project Consortium 2012), and Human Origins samples (Lazaridis et al. 2014); (2) an ALDER (Loh et al. 2013) analysis of the proportion and timing of African ancestry in these same populations; and (3) a SupportMix (Omberg et al. 2012) analysis of the population assignments of local genomic segments of the 96 Q1 (Bedouin), Q2 (Persian-South Asian), or Q3 (African) Qatari genomes. The ADMIXTURE analysis identified K = 12 ancestral populations as having the lowest cross-validation error (Supplemental Fig. 7A). At this level of resolution, the Q1 (Bedouin) had a high average (84%) proportion of ancestry that was also present in the Human Origins Bedouin B population at a high average proportion (93%) (Supplemental Fig. 7B,C), in which this same ancestry was also shared with Saudis, and at lower levels among other Middle Eastern populations. This ancestry therefore appears to be the signal of an indigenous Arab ancestral population. The Bedouin A population also shared this ancestry but at a lower average proportion (45%) and appeared to be more admixed overall. The Q2 (Persian-South Asian) shared a large proportion (45% on average) of ancestry that dominates in Iranians (46% on average), consistent with a Persian ancestral population (Omberg et al. 2012). The Q3 (African) shared the majority of ancestry with African populations as expected and were considerably admixed overall, again consistent with the known history of this subpopulation (Supplemental Fig. 7A; Omberg et al. 2012).  The ALDER analysis determined the relative percentage of African (Yoruba) ancestry in the Q1 (Bedouin) (2.6% ± 1.37) and Q2 (Persian-South Asian) (5.0% ± 1.41) at levels on par with estimates for other populations sampled in the region (Supplemental Fig. 8; Supplemental Table X), including Human Origins Bedouin and Saudi. This confirmed that recent African admixture is limited to the Q3 (African) subpopulation (37.6% ± 0.9), in which this estimate is on par with African American populations. An estimate of the timing of African admixture placed the number of generations for Q1 (Bedouin) (15.2) and Q2 (Persian-South Asian) (14.0) slightly higher than Q3 (African) (9.3), consistent with the Q1 (Bedouin) and Q2 (Persian-South Asian) reflecting more distant African admixture events and with the Q3 (African) reflecting the historical timing of the African slave trade in the region (Omberg et al. 2012). The SupportMix analysis used six of the 1000 Genomes populations (two European, two Asian, and two African) (see Supplemental Methods for details) as ancestral proxy reference panels and produced a set of “best guess” admixture assignments based on highest similarity to these genomes. Although these 1000 Genomes populations do not include appropriate local populations most closely related to the Qataris needed for assessment of the true admixture composition of the genomes, the ancestry track length distribution of haplotypes assigned to African populations (Yoruba or Luhuya) provides a qualitative indicator of whether the subpopulations experienced recent admixture with African populations. As expected, the track lengths of the Q1 (Bedouin) and Q2 (Persian-South Asian) assigned to African 1000 Genomes populations were far shorter than those for Q3 (African) (Supplemental Fig. 9), again confirming that recent African admixture is limited to the Q3 (African) subpopulation. |
|


