|
|
Post by Admin on Jan 21, 2023 19:16:38 GMT
Mobility in the Middle/Late Bronze Age Aegean For the LBA groups and the IA individual, we explored models of mixture from the corresponding ascending group (‘S. Mainland-Islands LN-EBA’ and ‘Crete EMBA’) and several European populations dated between around 3500 and 1000 BC (Supplementary Table 8). Informed by the previous analyses, we restricted the possible second sources to populations such as the EBA herders from the Pontic-Caspian Steppe (here grouped under ‘W. Eurasian Steppe En-BA’ and typically representing WES) and those shown to share a close genetic affinity with them. We first tested these models on ‘Site_Period’ groups, only if the cladality test (qpWave) agreed with grouping them as a homogeneous cluster (Supplementary Figs. 1 and 3a). Within the larger group from Chania, departures from cladality (P « 0.05) were more frequent (~10%) and were predominantly driven from specific individuals lying at the two ends of the EBA-LBA cline in the PCA (Extended Data Fig. 2b). To explore how these reflect significant differences in the admixture modelling, we analysed the group from Chania into the following three subgroups: ‘Chania LBA (XAN030)’, ‘Chania LBA (a)’ (XAN014, XAN028, XAN034) and ‘Chania LBA (b)’ (all the others) (Supplementary Table 8). We found various sources ranging from East Europe, to Central and South Europe adequately fitting most models for the LBA Aegean groups. The smaller and heterogeneous sample of BA Bulgarian individuals or BA Sicily did not fit. Models with Serbia (EBA), Croatia (MBA) and Italy (EMBA) were adequate most of the time, while those with ‘W. Eurasian Steppe En-BA’ (En, Eneolithic) or some Central European source (for example, Germany LN-EBA ‘Corded Ware’) were adequate for all groups at the P ≥ 0.01 cutoff. Therefore, at the moment it is not possible to more closely identify the region(s) from where this genetic affinity was derived. Among the groups of the southern mainland, the estimated coefficients of the WES-related ancestry are overlapping (±1 s.e.) and average to 22.3% (Fig. 4a) but were substantially lower than for Logkas in the northern mainland (43–55% ± 4%). No significant differences were noted for IA Tiryns (±1 s.e.), indicating—albeit with limited evidence—genetic continuity after the end of the BA at least for this site. Similar coefficient ranges as in the southern mainland are observed for the nearby islands and the Cyclades, although the model for the one individual from Salamis shows no WES-related ancestry. In sharp contrast, in Crete, WES-related coefficients vary from 0% to about 40% clustering in three groups with significantly different coefficients. Among the individuals with minimal/no WES ancestry are the earliest, dating to the late seventeenth or sixteenth century BC Aposelemis, whereas the youngest (Krousonas, Armenoi; twelfth century BC) harbour some of the highest amounts. However, within the ancient city of Chania, individuals spanning a short period of about three centuries display the entire range, a pattern consistent with an early phase of mixing between divergent populations. 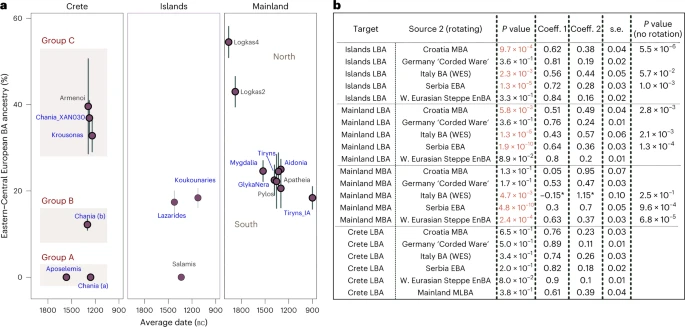 Fig. 4: Proximal two-way qpAdm models for the MLBA groups. a, Estimated mean coefficient (coeff.) (±1 s.e.) of additional ancestry (WES-related) using as proxy a BA Central European population (‘Germany LN-EBA Corded Ware’). For every group we assumed local ancestry in the models using the ascending population from the corresponding area (that is, EMBA Crete, LN-EBA southern Greek mainland and islands or LN northern mainland (for Logkas). Newly reported LBA groups are annotated in blue letters. Before we applied the modelling on every ‘Site_Period’ group, we performed a test of cladality among all individuals which suggested substructure within the LBA site of Chania in Crete and resulted in three analysis groups. Overall, individuals from LBA Crete are distributed in three groups of non-overlapping WES-related ancestry estimations (A, B and C). Models are supported with P ≥ 0.05, with the exception of Tiryns_IA and Pylos with P = 0.02 and 0.04, respectively. b, Modelling results using the approach of rotating competing sources 2 in the right populations set (R11) (Supplementary Note 2) for Crete, the mainland and the islands. Low P values (<0.01) indicate poor fit of the tested model and are annotated in red. For these models, the P values are compared with the model fit without rotation of the sources. The gradual shift in Crete can be explained with admixture from the mainland but other proximal sources fit equally well. P values and standard errors of mean were calculated by the qpAdm program applying a likelihood ratio test and the 5 cM block jackknifing method, respectively. No correction for multiple testing has been made. See also Extended Data Fig. 2 and Supplementary Tables 8 and 9. To better understand these remarkable ancestry patterns in LBA Crete, we tested competing admixture models by interchanging the candidate second sources in which we now included ‘Mainland MLBA’ that consisted of all the individuals from the third panel of Fig. 4a (both southern and northern). For a comparison, we also tested the same models on the grouped targets ‘Islands LBA’ (Euboea, Aegina, Salamis and Cyclades), ‘S. Mainland’ and ‘N. Mainland’—being aware that such artificial subdivisions of landscapes might not reflect past categorizations. The results are summarized in Fig. 4b. Interchanging the sources resulted in the rejection of some previously adequate sources (for example, ‘Italy BA’ for ‘Islands LBA’). Overall, proximal sources like EBA Serbia, MBA Croatia and BA Italy failed to model both mainland and island groups (P ≤ 5.80 × 10−3), whereas models with Central or Eastern European sources remained adequate. However, two-way models with all of the above sources as well as ‘Mainland MLBA’ fit the allele frequencies of all the LBA individuals from Crete (‘Crete LBA’). This also applied when we modelled the two clusters from LBA Crete separately (Fig. 4a and Supplementary Table 9) but for the Crete LBA (group C) with high WES ancestry (individuals XAN030, KRO008, KRO009 and published Armenoi), just one source from ‘Mainland MLBA’ became adequate. |
|
|
|
Post by Admin on Jan 22, 2023 21:33:11 GMT
Insights into sex bias, biological kinship and marital practices Studies have shown that in some regions of Europe—like the Iberian Peninsula, Central Europe and Britain—the large-scale gene flow associated with the Eurasian Steppe during the BA resulted in the prevalence of the Y chromosome R1a and R1b haplogroups28 or even involved male-biased admixture33,39,40. For the Aegean, we also estimated a significantly lower WES-ancestry proportion on the X chromosomes of the male individuals compared to most of the autosomes, which is consistent with male-biased admixture (Extended Data Fig. 3). However, only four out of the 30 male individuals dating post-sixteenth century BC (LBA and IA) carry the R1b1a1b Y haplogroup. The remaining—as well as the EBA/MBA ones—attest to the high prevalence of Y haplogroups J and G/G2 (39 and 10 out of 59, respectively; Supplementary Table 2). These were already present in Early Holocene Iran/Caucasus and among Anatolian and European farmers41,42,43,44,45 and very common in the Chalcolithic Anatolia and the Levant as well42,46,47, further highlighting the importance of the contacts between the Aegean and southwest Asian populations since the Early Neolithic. Biological relatedness and its representation in prehistoric collective burials has been poorly understood in the Aegean. Here, we present the first evidence for representation of biologically kin groups from a collective intramural infant grave dating to the LBA—a type of burial which existed since the Neolithic Aegean but became more common since the MBA48,49. Located within the Mycenaean (LBA) settlement in Mygdalia, a small cist grave was the primary inhumation of at least eight perinatal infants and one of the six child burials under the houses of the settlement (Supplementary Note 1). By estimating the degree of relatedness among seven of these infants (Methods; Extended Data Fig. 4 and Supplementary Note 3) and assigning the uniparental haplogroups (Supplementary Table 2), the relationship of the infants could be resolved in a single extended family tree whereby the six infants were the children and grandchildren of one couple (Fig. 5). The seventh individual (MYG004) was not a direct offspring of this family but related to MYG005 in the third degree through the maternal line, plausibly as first cousins. Fig. 5: Reconstruction of the family tree for the infants from the burial in Mygdalia (MYG; solid colour shapes). 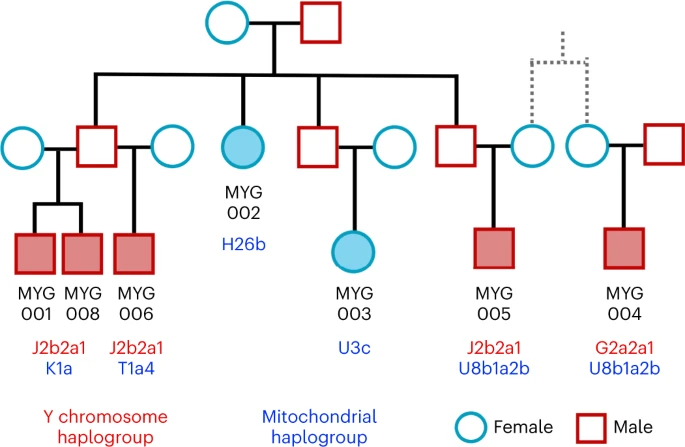 Additional evidence of biological relatedness comes from Aidonia, where pairs of first- to third-degree relatives were determined among individuals buried within the three chamber tombs and the ossuary of Hagios Charalambos at the Lasithi plateau (Supplementary Note 1 and Extended Data Fig. 4). The individuals studied from Hagios Charalambos represent a secondary deposition of intermingled skeletons but were all unearthed from a particular section of the cave (Supplementary Note 1). Besides some pairs of close relatives (first to second degree), many pairs represent distant relatives. In addition to this high frequency of distant genetic relatedness, we also report extraordinarily high levels of consanguinity (~50% of the 27 individuals) estimated from the runs of homozygosity (ROH) by performing hapROH on the genotyping data50 (Fig. 6a; Methods). The individual ROH histograms matched more with the expectations for parents being related to the degree of first cousins, half-siblings and aunt/uncle–nephew/niece (Extended Data Fig. 5). However, given the stochastic nature of genetic recombination and the often-compromised coverage of ancient samples, one individual’s genome might only noisily match the expectations. Therefore, we combined the possible first-cousins unions cases and the cumulative histogram this produced favoured the parental relationship of first cousins against other scenarios (Fig. 6b and Extended Data Fig. 6). Coupling the evidence for frequent distant relatives and cousin–cousin unions suggests that those individuals formed a small endogamous community that regularly practiced first-cousin intermarriages. |
|
|
|
Post by Admin on Jan 23, 2023 20:48:15 GMT
Fig. 6: Runs of homozygosity estimated with hapROH. 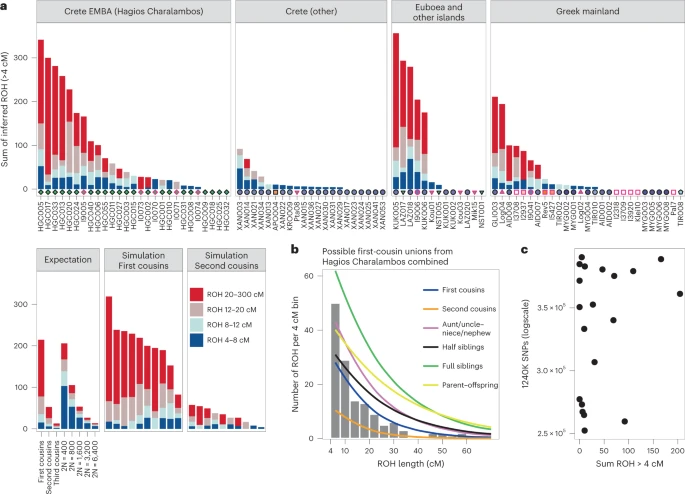 a, Inferred ROH per ancient Aegean individual. Results are plotted by area and the archaeological period/date of each individual is provided following the same symbol/colour scheme introduced in Fig. 1. Simulations and expectations for given parental relationships and demographic scenarios are given. For many individuals the ROH length distribution matches close-kin unions (first and second cousins). b, Combined histogram of ROH length from all close-union offspring cases from the ossuary of Hagios Charalambos at the Lasithi plateau in Crete, compared to expected densities for certain parental relationships. See also Figs. 5 and 6. c, Scatterplot of lower coverage samples (250,000–400,000 SNPs) with total length of inferred ROH indicates that hapROH can reliably estimate long ROH at lower thresholds (Methods). Intriguingly, endogamy is not a unique feature of Hagios Charalambos. We applied the method on another 61 Aegean individuals from all the periods that met recommended SNP coverage thresholds. In total, we found that ~30% of the individuals have most of their ROH in the bin of the longest ROH blocks, consistent with being offspring of parents related to a degree equivalent to first and second cousins (Fig. 6a). Offspring of close-kin unions were identified from the Neolithic through the LBA but due to the uneven sampling no conclusions can be drawn regarding temporal trends. Consanguinity was also present in higher frequency in the smaller islands of Salamis, Lazarides, Koukounaries and Koufonisia (50%) but overall it seemed common throughout the Aegean. The observed high frequency of endogamy diachronically points to a rather common social practice in the prehistoric Aegean that is so far unattested in the rest of the global aDNA record50. Finally, we observe a lowered genetic diversity among the Neolithic Aposelemis individuals, measured by a substantially reduced rate of mismatching alleles between pairs of samples (median P0 ≈ 0.22) (Extended Data Fig. 4; Methods). This signal can be due to several reasons. First, a lower P0 would be consistent with Aposelemis being a small endogamous community; however the absence of any long ROH in APO004, the single individual with sufficient coverage to infer long ROH, does not support this hypothesis. Second, the lowered pairwise diversity could represent multiple pairs of second-degree relatives. However, to fit all pairs into a single consistent pedigree would require that all six individuals are half-siblings from either the maternal or the paternal side, with the exception of a single pair of full siblings (APO004–APO028). Due to the low SNP coverage in all the individuals, uniparental markers can neither rule out nor confirm such a pedigree but its high specificity places it as a less likely scenario. Finally, long-term reduction of population size (bottlenecks) can cause lower population heterozygosity and such a signal has been previously reported for instance in hunter-gatherer groups and Cardial Neolithic Iberians51,52. Individuals from such drifted populations are expected to exhibit shorter ROH (4–8 cM), which are currently not detectable in low-coverage individuals such as APO004. Further supporting this scenario, the inferred heterozygosity (h) within the Aposelemis individuals was also reduced (mean h ≈ 0.1) and close to the expectation when assuming that the average pairwise diversity (P0 ≈ 0.2) represents the diversity of the population and not pairs of close relatives. Summarizing, the current evidence is most consistent with the scenario of the Aposelemis early farmers descending from a small-sized population. |
|
|
|
Post by Admin on Jan 24, 2023 21:08:55 GMT
Discussion
Our large-scale archaeogenomic approach provides new evidence regarding the role of human mobility in Aegean prehistory. The unprecedented finding of high frequency of consanguinity reveals a cultural practice otherwise unattested in the archaeological record.
First, our analyses on the Neolithic cemetery of Aposelemis, postdating the earliest levels at Knossos by about 1,000 years, suggest an Anatolian origin of the first Neolithic settlers, consistent with architectural, palaeobotanical and lithic evidence53 and recent evaluation of wild and domestic fauna at those earliest levels that also suggest animal husbandry54. While a similar genetic connection was observed for coeval mainland populations24,25, the genetic impact of Mesolithic and Neolithic populations from the other Aegean Islands, remain unknown but the evidence of a pre-Neolithic island horizon of a seafaring tradition55 forces us to further elucidate the role of hunter-gatherers in the uptake of Neolithic subsistence practices in future studies. Thus, the reduced heterozygosity of the Aposelemis population might be interpreted as a coalescence of a small population of Anatolian farmers who settled the island in the early seventh millenium BC and remained biologically isolated for a period of time or as mobile small-sized populations arriving from nearby islands or a combination of both.
Subsequently, our findings indicate that the genetic landscape of Crete changed substantially since the early sixth millenium BC, marked by an influx of Anatolian populations inferred with our qpAdm modelling and admixture dating. Interestingly, eastern gene flow is also evident in other parts of Greece (Euboea, Aegina and Cyclades) since the LN but seems more episodic and oriented to populations from the Caucasus. In addition, although Y haplogroups are unresolved, male exogamy should be discussed as a plausible contributing factor to the heterogenous genetic profiles among the male individuals from Nea Styra, in line with evidence from biodistance on a neighbouring site35. Overall, while a more even sampling would be critical, current data seem to support that the eastern gene flow was not symmetric across the Aegean.
The disruption of life that is manifested in the Aegean and the Balkans via settlement dislocation during the late third millenium BC could be related to a breakdown of social structures and/or climatic challenges56. The finding of ‘northern’ ancestry in the MBA and LBA populations from the Greek mainland, does not support a large-scale population displacement but the north–south gradient indicates the directionality of this migration and population mingling. Some putatively proximal sources like ‘Serbia EBA’ or ‘Bulgaria BA’ failed to model this ‘incoming’ ancestry in many groups and R1b Y haplogroups were rather infrequent among LBA Aegean groups, all of which points to different migration dynamics in the BA Balkans and Greece, compared to other parts of Central and Western Europe.
A more direct demographic connection can be proposed regarding the LBA Cretan and Greek mainland populations. Following an horizon of destructions targeting palatial centres and elite symbols in Crete (Late Minoan IB)57, material culture, funerary architecture and burial practices are believed to express innovations with features traditionally ascribed to the Mycenaean culture. On these grounds, an invasion of the island by people from the Greek mainland (around fifteenth century BC) has been proposed but remains highly contested12,58,59,60. While unable to settle this debate decisively, the genetic analyses demonstrate that Cretan populations at larger port cities biologically mixed with populations coming to the island during the course of a few centuries. The presence of individuals with some of the highest WES-related ancestry proportions within LBA Aegean (Crete LBA group C), despite fitting with a scenario that the Greek mainland was the only source of incoming people, it could also suggest that populations from more distant areas (for example, Italy) contributed to the Crete LBA transition, a possibility that is supported in the material culture as well61,62,63.
All different migrations proposed here (to Crete during the Neolithic and EBA, to the Greek mainland before the LBA and from the mainland to Crete during the LBA) differ in their bioarchaeological evidence, which, therefore, must not be seen as a simple proof of an archaeological hypothesis but as an additional perspective enabling us to unravel the complexity of past mobilities.
Finally, the evidence for consanguinity adds another layer regarding human mobility and social practices. Since the fundamental work by ref. 64, the phenomenon of cross-cousin unions has been much debated in anthropology, whereby in present-day societies, the evidence for cross-cousin unions is diverse, ranging from a common practice via toleration up to prohibition65. Different social, economic and ecological arguments have been brought forward as underlying reasons, for example, geographic isolation, endemic pathogen stress, integrity of inherited land and so on66. A combination of several factors combined with subsistence-specific needs (for example, olive cultivation forcing local constancy) might have shaped this practice in the Aegean. However, small population size was probably not a major reason in the Aegean as the reduced short-range ROH shown in our analyses is consistent with larger population sizes. Moreover, cross-cousin unions were practiced in different geographic contexts—on islands of different sizes as well as the Greek mainland and are not evident at some places during the second millennium (for example, Chania). Future studies need to further elucidate the factors that were responsible for the emergence, continuity and disappearance of marital practices. So far, the importance of cross-cousin unions in the prehistoric Aegean is unique among the currently available data for prehistoric endogamy, which is otherwise rarely evidenced50,67,68,69. This might indicate different standpoints with respect to marital practices of rural versus urban societies and/or that those were amenable to cultural influences and changed over time. Studying the interplay between past mortuary practices and social structure—including marital or residence rules—from an integrative bioarchaeological perspective has just become possible and future studies will help to refine our understanding of past social belonging.
|
|
|
|
Post by Admin on Jan 26, 2023 17:54:46 GMT
Methods No statistical methods were applied for the determination of sample size and randomization. The overall burial record from the Aegean Bronze Age is a corpus which underwent specific selection criteria in the past and has been subject to specific modes of preservation and excavation since then (for example, only individuals with a certain status and/or age were buried in a way that allows their study at present). The corpus of samples analysed in this study represents a broad variety of burial contexts (for example, shaft graves/collective graves, single graves, primary and secondary burials) through time and none of the burials would be termed ‘elite’ or ‘outstanding’ in its respective archaeological/historical context. There is also no sampling bias with respect to sex, age or locality of the burials and diverse cultural settings were included (for example, individuals from urban centres like Tiryns and Chania and remote hamlets like Mygdalia). Preparation of aDNA analysis For the purpose of this study, we sampled 385 skeletal elements originally assigned to 357 ancient individuals. Teeth and petrous bones made >95% of the sample corpus but when these elements were missing other parts such as tibia and femora were chosen. All sampling took place in a dedicated aDNA laboratory of MPI-SHH in Jena, following the laboratory’s archived protocols doi.org/10.17504/protocols.io.bqebmtan and doi.org/10.17504/protocols.io.bdyvi7w6, the latter being an adaptation of a published protocol70. The aDNA extraction from most of the bone powder samples was performed with a modified silica-based protocol71. A detailed description of the steps is given in doi.org/10.17504/protocols.io.baksicwe. Genomic libraries were prepared from these extracts according to a double-stranded (ds) library protocol72 with an initial step of partial UDG treatment73 (https://doi.org/10.17504/protocols.io.bmh6k39e), followed by Illumina dual indexing (https://doi.org/10.17504/protocols.io.bakticwn). For a portion of the samples, we used an extraction-to-indexed library protocol supported by an automated liquid-handling system74,75 which constructs libraries from single-stranded (ss) molecules. From every extract, at least one of the produced libraries was initially sequenced at a low depth (5–10 million reads) on an Illumina HiSeq400 platform with a setup of 50 cycles and paired-end or 75 cycles and single-read sequencing. Raw FastQC files were processed through EAGER pipeline76 for removal of adaptors (AdapterRemoval v.2.2.0; ref. 77), mapping against the human reference hs37d5 with the Burrows–Wheeler aligner (BWA; v.0.7.12; ref. 78) with mapping quality and length filters of 30, and removal of PCR duplicates with dedup (v.0.12.2; ref. 76). Resulting information about library complexity and percentage of endogenous DNA was combined with mapDamage (v.2.0.6; ref. 79) estimates to evaluate the profile of endogenous aDNA preservation (Supplementary Table 1). Overall, our initial screening revealed that human aDNA preservation was very low to moderate (0.1–10% human endogenous DNA). Therefore, only aDNA enrichment methods are an economically viable strategy that allows one to generate data from a large number of individuals. Here, we chose to minimize batch effects and consistently generated in-solution hybridization enrichment data, consisting of ~1,2 million ancestry-informative positions (1240K capture)28,43,80,81 from all samples with 0.1% human endogenous DNA or more. We note that a small proportion of the sampled libraries exhibited high DNA preservation (nine samples with >10% and up to ~40% endogenous content), which would make sequencing of the whole human genome cost-efficient and doing so could address additional research questions (for example, about rare variants). Only part of the immortalized libraries was used to produce enrichment data. The remaining libraries are permanently stored at the MPI-SHH/EVA laboratory facilities and future studies can use this resource to generate whole-genome data from these libraries. Following the 1240K enrichment, the selected libraries were sequenced at standard ~20 million reads. For the evaluation of the post-1240K capture data, we rerun EAGER and mapDamage with the same settings. We also used the bed file of 1240K SNP positions to estimate on-target endogenous before-and-after 1240K capture and evaluate the performance of the protocol. We used Preseq (v.2.0; ref. 82) with the parameters <lc_extrap -s 1e5 -e 1e9> to predict the unique reads yielded in larger sequencing experiments. For libraries with low complexity, whenever that was possible, we opted for preparation of multiple libraries from the same extract. Additional sequencing data from the same library or multiple libraries from one DNA extract or same individual that were produced with the same protocols were processed equally and all data were merged at the level of bam files with Samtools (v.1.3) and dedup was run again. We authenticated aDNA using three different methods on the bam files that estimate modern DNA contamination on ancient samples. We analysed single-stranded, no-UDG-treated libraries with AuthentiCT (v.1.0.0; ref. 83) that relies on the distribution of damage-induced deamination across the length of the ancient molecules. We run the module for contamination estimate on males from ANGSD84, which relies on heterozygosity on polymorphic SNPs on the X chromosome. We previously trimmed bams for terminal damage with trimBam (https://genome.sph.umich.edu/wiki/BamUtil:_trimBam) and reported the method 1 estimation. Finally, we analysed all libraries with schmutzi85 after mapping mitochondrial reads with CircularMapper (v.1.93.5) and removing duplicates76 and downsampling to 30,000 reads. Run modules contDeam and schmutzi estimated endogenous deamination, called an endogenous consensus and, based on this, computed the contamination rate. Ratios of mitochondrial/nuclear DNA that are very high (>200) can be unreliable for mitochondrial contamination estimates86. Therefore, when applicable, we relied on other methods and/or the behaviour of such samples in population genetic analyses. The genetic sex was determined from a scatterplot of coverage on X and Y chromosomes normalized for autosomal coverage, which provided an unambiguous distinction between males and females and also matched the macroscopic estimations for adult individuals in all but a few exceptions (Supplementary Note 1). We extracted genotypes from the pileups of original and trimmed bam files of ds libraries using the tool pileupCaller (https://github.com/stschiff/sequenceTools/tree/master/src/SequenceTools) and the option randomHaploid, which randomly chooses an allele to represent the genotype at a given SNP position. For the final genotype file, we kept transitions from the masked version and transversions from the original version. We genotyped the pileups from ss-library bams by activating the option singleStrandMode in pileupCaller which filters out forward-mapping reads with a C-T polymorphism and reverse-mapping reads with a G-A polymorphism, thereby effectively removing bias due to damage. Because of the differences in data production between ds and ss libraries, when applicable, we merged such libraries on the genotype level by randomly choosing a non-missing genotype at every position. Individuals with <20,000 SNPs, ≥10% contamination estimate or absence of such estimate were excluded from subsequent analyses. For selected individuals, we run pileupCaller with the option -randomDiploid and calculated within individual heterozygosity as number of ht sites/number of all sites. We merged our final dataset with the release of publicly available genotype datasets of ancient and modern individuals (v.50.0) (https://reich.hms.harvard.edu/allen-ancient-dna-resource-aadr-downloadable-genotypes-present-day-and-ancient-dna-data), to which we added the recently published aDNA data from Italy87 and based our inferences on a subset of the published data older than 2,000 years from across Eurasia. For the merging with the worldwide modern populations on the Human Origins array (~0.5 million SNPs) we kept the intersection of SNPs between the two panels. For downstream analyses we restricted all data to the 22 autosomes. We assigned mitochondrial haplogroups and haplotypes from the consensus sequence (q30) generated by schmutzi and the software Haplogrep (v.2.1.25; ref. 88) applying a quality threshold of 0.65. To assign Y haplogroups, we filtered the pileup from the trimmed bams for ISOGG SNPs and for every such SNP we calculated its record of being either ancestral or derived. Then, via manual inspection we checked whether the presence of diagnostic SNPs for a given haplogroup followed a root-to-tip trajectory or whether there were spurious jumps in the phylogeny because of damage. For libraries with low coverage on mitochondrial and Y chromosome DNA, we additionally performed whole-genome and SNP enrichments, respectively, according to established protocols81,89. A summary of genetic sex, contamination estimates, SNP coverage and Y/mito-haplogroup assignments is given in Supplementary Table 2. |
|

