|
|
Post by Admin on Aug 19, 2023 18:58:13 GMT
Natural selection and SARS-CoV-2 responses To investigate the contribution of natural selection to population differences in immune responses, we first searched for overlaps between (r)eQTLs and genome-wide signals of local adaptation, measured by the population branch statistic (PBS)39. We identified 1,616 eQTLs (1,215 genes) and 180 reQTLs (166 genes) displaying strong population differentiation (empirical P < 0.01), 90 of which were ancestry specific (Supplementary Table 7a and Supplementary Note 6). Among genes harbouring putatively adaptive (r)eQTLs, we found key players in IFN-mediated antiviral immunity, such as DHX58 and TRIM14 in Africans, ISG20, IFIT5, BST2 and IFITM2-3 in Europeans, and IFI44L and IFITM2 in East Asians. We then used CLUES40 to identify rapid changes in (r)eQTL frequency over the last 2,000 generations (that is, 56,000 years) in each population (Supplementary Fig. 9 and Supplementary Table 7b). We found signals of rapid adaptation (maximum |Z| > 3) targeting the same (IFITM2, IFIT5) or different (ISG20, IFITM3, TRIM14) eQTLs at highly differentiated genes, suggesting repeated adaptations targeting IFN-mediated antiviral immunity (Supplementary Note 10, Supplementary Table 7c and Supplementary Fig. 10). We determined whether selection had altered gene expression in specific cell types or in response to SARS-CoV-2 or IAV by testing for increased population differentiation (PBS) at (r)eQTLs within each cell type, relative to random single-nucleotide polymorphisms (SNPs) matched for allele frequency, linkage disequilibrium (LD) and distance to the nearest gene. In the basal state, eQTLs were more strongly differentiated in Europeans, the strongest signal observed for γδ T cells (Extended Data Fig. 6a). Among popDEGs for which genetics mediates more than 50% of the differences between Africans and Europeans, 34% presented signals of rapid adaptation in Europeans (versus 21% in Africans, Fisher’s exact test, P = 7.7 × 10−6). For example, population differences at GBP7 have been driven by a frequency increase, over the last 782–1,272 generations, of the rs1142888-G allele in Europeans (maximum |Z| > 4.3, Extended Data Fig. 6b). Focusing on responses to viruses, SARS-CoV-2 reQTLs displayed increased population differentiation in East Asians (FE = 1.24, one-sided resampling, P < 2 × 10−4; Extended Data Fig. 6c) and were enriched in East-Asian-specific variants (OR > 4.2, Fisher’s exact test, P < 2.3 × 10−6; Supplementary Note 6 and Supplementary Table 7d). Furthermore, among SARS-CoV-2-specific reQTLs, 28 reQTLs (5.3%) displayed signals of adaptation in East Asians starting 770–970 generations ago (around 25,000 years)—a timeframe associated with genetic adaptation at SARS-CoV-2-interacting proteins23 (OR relative to other populations = 2.6, Fisher’s exact test, P = 7.3 × 10−4; Fig. 4a and Extended Data Fig. 7a–c). An example is the immune mediator LILRB1, which has a SARS-CoV-2-specific reQTL (rs4806787) in pDCs (Extended Data Fig. 7d). However, the selection events making the largest contribution to the differentiation of SARS-CoV-2 responses in East Asia (top 5% PBS) began before this period (more than 970 generations ago, OR = 1.94, Fisher’s exact test, P = 0.019; Fig. 4b). For example, the rs1028396-T allele (80% frequency in East Asia versus 16–25% elsewhere), associated with a weaker response of SIRPA to SARS-CoV-2 in CD14+ monocytes, presents a selection signal beginning more than 45,000 years ago (Fig. 4b and Extended Data Fig. 7e). SIRPα inhibits infection by endocytic viruses, including SARS-CoV-241. These results suggest recurrent genetic adaptation targeting antiviral immunity over the last 50,000 years, contributing to present-day population differences in immune responses to SARS-CoV-2. Fig. 4: Natural selection effects on population differentiation of immune responses. 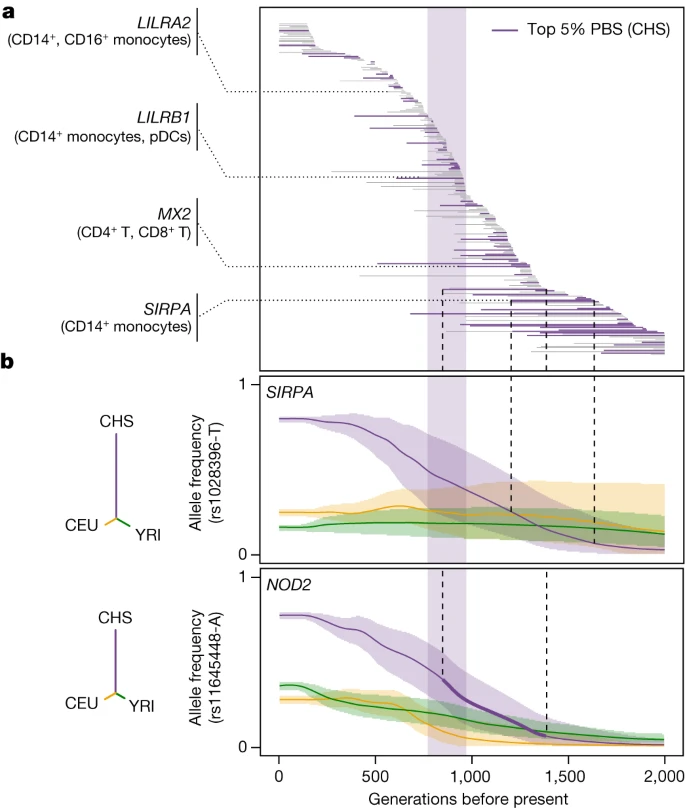 a, Estimated periods of selection, over the past 2,000 generations, for 245 SARS-CoV-2 reQTLs with significant signals of rapid adaptation in East Asians (CHS) (maximum |Z| > 3). Each horizontal line represents a variant, sorted in descending order of time to onset of selection. The area shaded in purple highlights the period (770–970 generations ago) associated with genetic adaptation at host coronavirus-interacting proteins in East Asians23. Several immunity-related genes are highlighted. b, Allele frequency trajectories of two SARS-CoV-2 reQTLs (rs1028396 at SIRPA and rs11645448 at NOD2) in Africans (YRI, green), Europeans (CEU, yellow) and East Asians (CHS, purple). The full lines indicate the maximum a posteriori estimate of allele frequency at each epoch and shaded areas indicate the 95% CIs. The dendrograms show the estimated unrooted population phylogeny for each eQTL based on PBS (that is, the branch length between each pair of populations is proportional to −log10[1 − FST]). |
|
|
|
Post by Admin on Aug 20, 2023 19:48:28 GMT
Contribution of eQTLs to COVID-19 risk We investigated the contributions of genetic variants altering responses to SARS-CoV-2 ex vivo to COVID-19 risk in vivo by determining whether (r)eQTLs were more strongly associated with COVID-19 GWAS hits8 than random, matched SNPs (Methods). We observed an enrichment in eQTLs at loci associated with susceptibility (reported cases) and severity (hospitalized or critical cases) (FE = 4.1 and FE > 3.8, respectively, one-sided resampling, P < 10−4), and a specific enrichment in reQTLs at severity loci (FE > 3.7, one-sided resampling, P < 3 × 10−3; Fig. 5a). This trend was observed across most cell lineages (Extended Data Fig. 10a). Colocalization analyses identified 40 genes at which there was a high probability of (r)eQTL colocalization with COVID-19 hits (posterior probability that both traits are linked to the same SNP (PPH4 ) > 0.8) and transcriptome-wide association studies (TWASs) linked predicted gene expression with COVID-19 risk for 30 of these genes (FDRTWAS < 0.01; Supplementary Table 9a). These included direct regulators of innate immunity, such as IFNAR2 in non-stimulated CD4+ T cells, IRF1 in non-stimulated NK and CD8+ T cells, OAS1 in lymphoid cells stimulated with SARS-CoV-2 and IAV, and OAS3 in SARS-CoV-2-exposed CD16+ monocytes (Fig. 5b and Extended Data Fig. 10b,c). These results support a contribution of immunity-related (r)eQTLs to COVID-19 risk. Fig. 5: eQTLs and reQTLs contribute to COVID-19 risk. 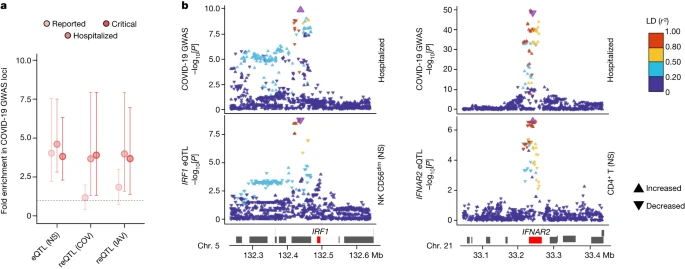 a, Enrichment in GWAS loci associated with COVID-19 susceptibility and severity at eQTLs and reQTLs. Data are the mean and 2.5th–97.5th percentiles (95% CIs) of fold enrichments observed over n = 10,000 resamplings. b, Colocalization of IRF1 and IFNAR2 eQTLs with COVID-19 severity loci. Top, the −log10 profiles (two-sided Student’s t-tests) for association with COVID-19-related hospitalization. Bottom, the −log10 profiles for association with expression in non-stimulated CD56dim NK cells (IRF1) and CD4+ T cells (IFNAR2). The colour code reflects the degree of LD (r2) with the consensus SNP identified by colocalization analyses (purple). For each SNP, the direction of the arrow indicates the direction of the effect. Chr., chromosome.
Focusing on the evolutionary factors affecting COVID-19 risk, we identified 20 eQTLs that (1) colocalized with COVID-19 hits (PPH4 > 0.8) and (2) presented positive selection signals (top 1% PBS, n = 13 eQTLs) or evidence of archaic introgression (n = 7 eQTLs), 14 of which regulate genes of which the expression is correlated with COVID-19 susceptibility and/or severity (FDRTWAS < 0.01) (Fig. 6). For example, two variants in high LD at DR1 (rs569414 and rs1559828, r2 > 0.73) displayed extremely high levels of population differentiation, probably due to selection outside Africa (DAF = 0.13 in Africa versus higher than 0.62 in Eurasia; Extended Data Fig. 10d). DR1 suppresses type I IFN responses49 and the selected alleles, which decrease COVID-19 severity, reduce DR1 expression in most immune cells (Fig. 6). Likewise, an approximately 39 kb Neanderthal haplotype, spanning the MUC20 locus in Eurasians, contains the rs2177336-T allele that increases MUC20 expression in SARS-CoV-2-stimulated cells, particularly for CD4+ T cells, and decreases COVID-19 susceptibility (Fig. 6). Together, these results reveal how past selection or Neanderthal introgression have impacted immune responses that contribute to present-day disparities in COVID-19 risk.
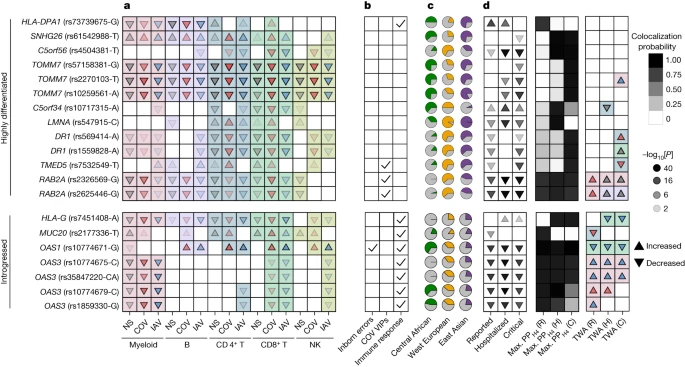
Fig. 6: Adaptation and archaic introgression at COVID-19-associated (r)eQTLs.
a–d, Features of (r)eQTLs colocalizing with COVID-19 risk loci (PPH4 > 0.8) and presenting either strong population differentiation (top 1% PBS genome-wide) or evidence of Neanderthal introgression. a, Effects of the target allele on gene expression across immune lineages and stimulation conditions. b, Clinical and functional annotations of associated genes. c, Present-day population frequencies of the target allele. d, The effects of the target allele on COVID-19 risk (infection, hospitalization and critical state), colocalization probability and the lineage and condition in which gene expression most likely affects COVID-19 risk as detected by transcriptome-wide association (TWA) analyses. For expression or COVID-19 associations, the arrows indicate increases/decreases in expression or disease risk with each copy of the target allele, and the opacity reflects the strength of association (two-sided Student’s t-test −log10 ). For the TWA analysis, the arrows indicate the effect of an increase in gene expression on the risk of COVID-19. In a and d, the arrow colours indicate stimulation conditions (non-stimulated (grey), SARS-CoV-2-stimulated (red), IAV-stimulated (blue)) and the background colour indicates the lineage (myeloid (pink), B (purple), CD4+ T (blue), CD8+ T (green), NK (light green)). For each eQTL, the target allele is defined as (1) the derived allele for highly differentiated eQTLs or (2) the allele that segregates with the archaic haplotype for introgressed eQTLs. When the ancestral state is unknown, the minor allele is used as a proxy for the derived allele. Note that, in some cases (for example, OAS1), the introgressed allele can be present in Africa, which is attributed to the reintroduction in Eurasia of an ancient allele by Neanderthals46. C, critical; H, hospitalized; R, reported.
|
|
|
|
Post by Admin on Aug 21, 2023 20:13:15 GMT
Discussion
Here we show that cell type composition is a major driver of population differences in immune responses to SARS-CoV-2. The higher proportions of memory cells in lymphoid lineages from individuals of African descent, along with their association with CMV infection, highlight how previous environmental exposures can contribute to population disparities in cellular activation states. Neglecting socioenvironmental factors that covary with ancestry may therefore inflate the estimated effects of genetic ancestry on phenotypic variation. One such factor is CMV, affecting leukocyte responses to SARS-CoV-2, but the impact of other exposures on population variation in immune responses remains to be determined. Common genetic variants can also contribute to immune response variation, but their effects primarily apply to a subset of genes showing strong population differentiation. This is illustrated by the rs1142888-G allele, which accounts for the greater than 2.8-fold higher levels of GBP7 expression in response to viral stimulation in Europeans compared with in Africans. The higher frequency of this allele in Europe probably results from selection occurring 21,900–35,600 years ago. GBP7 facilitates IAV replication by suppressing innate immunity50, but also regulates host defence to intracellular bacteria such as Listeria monocytogenes and Mycobacterium tuberculosis51, providing a plausible mechanism for positive selection at this locus.
This study also shows that natural selection and Neanderthal introgression contributed to differentiate present-day immune responses to SARS-CoV-2. We found traces of selection targeting SARS-CoV-2-specific reQTLs around 25,000 years ago in the ancestors of East Asians, coinciding with the proposed timing of an epidemic that affected the evolution of host coronavirus-interacting proteins23,24. However, there is little overlap between alleles selected during this period and variants underlying COVID-19 risk, suggesting changes in the genetic basis of infectious diseases over time, possibly due to the evolution of viruses themselves. Nevertheless, we identified cases (for example, DR1, OAS1-3, TOMM7, MUC20) in which selection or archaic introgression contributed to changes in both SARS-CoV-2 immune responses and COVID-19 outcome. Studies based on ancestry-aware polygenic risk scores from cross-population GWAS will be required to establish a formal link between past adaptation and present-day population differences in COVID-19 risk.
Finally, the genetic dissection of variation in transcriptional responses to SARS-CoV-2 provides mechanistic insights into the effects of alleles that are associated with COVID-19 risk. Variants of IRF1, IFNAR2 and DR1 associated with lower COVID-19 severity increase type I IFN signalling in lymphoid cells by upregulating IRF1 and IFNAR2 or downregulating DR1, attesting to the importance of efficient IFN signalling for a favourable clinical outcome4,12,13,14. Another example is MUC20, at which we identified a Neanderthal-introgressed eQTL that increases MUC20 expression in SARS-CoV-2-stimulated CD4+ T cells and decreases COVID-19 susceptibility. Given the role of mucins in forming a barrier against infection in the respiratory tract, the high MUC20 expression in ciliated epithelial cells from the bronchus52 and the detection of the MUC20 eQTL in pulmonary tissue (Supplementary Note 11), we suggest that the greater resistance to infection conferred by the Neanderthal haplotype may result from a similar effect on MUC20 expression in the respiratory tract.
We note two main limitations of our results. First, our samples mostly originate from male individuals, so the impact of sex on immune variation was not addressed. Sex has a widespread yet moderate effect on both transcriptional responses to microbial threats53 and the genetic regulation of gene expression54, supporting the transferability of our main conclusions. Nonetheless, examining sex-balanced cohorts will enable the characterization of possible sex-specific differences at the population scale. Second, given the sample sizes and cell counts needed to accurately define population variation in immune activity, we focused on a single system (PBMCs) and selected viral strains. Although PBMCs constitute a valuable model to characterize peripheral immune activation by SARS-CoV-29,10, they provide an incomplete representation of the pulmonary epithelium—the primary infection site for respiratory viruses. However, we found that 38% of the eQTLs identified in this study are also detected in lung tissue55, rising to 72% for eQTLs shared across immune lineages (Supplementary Note 11 and Supplementary Table 9b). Further studies are needed to examine the transferability of our findings to other cell types and to investigate how diverse viral strains affect the dynamics of host responses to SARS-CoV-2.
Overall, our results highlight the value of single-cell approaches in capturing the full diversity of peripheral immune responses to RNA viruses, particularly SARS-CoV-2, and provide insights into environmental, genetic and evolutionary drivers of immune response variation across individuals and populations.
|
|
|
|
Post by Admin on Aug 26, 2023 19:11:37 GMT
SARS-CoV-2: How the history of human populations influences their immune response by Pasteur Institute 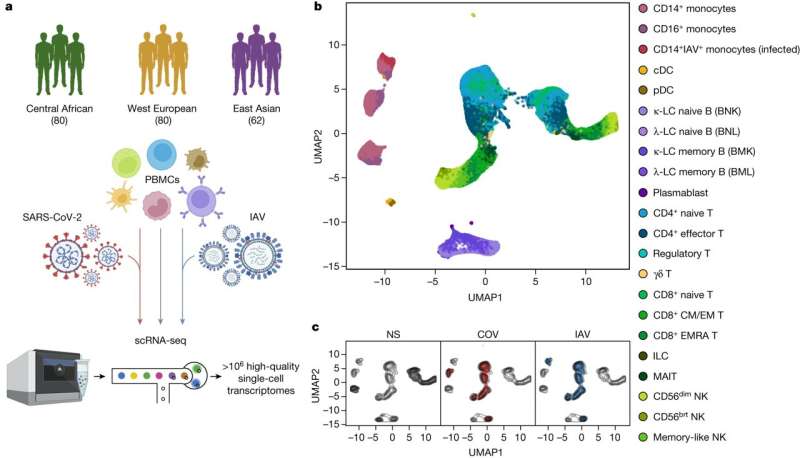 During the COVID-19 pandemic, the clinical spectrum observed among people infected with SARS-CoV-2 ranged from asymptomatic carriage to death. Researchers at the Institut Pasteur, the CNRS and the Collège de France, in collaboration with researchers around the world, have investigated the extent and drivers of differences in immune responses to SARS-CoV-2 across populations from Central Africa, Western Europe and East Asia. They show that latent cytomegalovirus infection and human genetic factors, driven by natural selection, contribute to population differences in immune response to SARS-CoV-2 and the severity of COVID-19. Understanding the factors underlying such population disparities could help to improve patient management in future epidemics. These results were published on August 9, 2023 in Nature. The Institut Pasteur's Human Evolutionary Genetics Unit, led by Lluis Quintana-Murci, investigates how human populations differ in their immune responses to infection. These differences may result from different environmental exposures or from past population history, including natural selection, shaping the patterns of genetic diversity of human groups. In this study, the scientists investigated the extent and causes of disparities in the responses to the SARS-CoV-2 virus, focusing on populations from different geographic and ethnic backgrounds. During the COVID-19 pandemic, the SARS-CoV-2 virus caused a wide range of clinical manifestation, from asymptomatic infection to fatal disease. Although advanced age remains a primary risk factor, male gender, comorbidities and various human genetic and immunological factors also contribute to disease severity. To study variations in immune responses to SARS-CoV-2 across human populations, scientists exposed immune blood cells from 222 healthy donors from Central Africa, Western Europe, and East Asia to the virus. Single-cell RNA sequencing was used to analyze the SARS-CoV-2 responses of 22 blood cell types. These data were then combined with serological and genetic information collected from the same individuals, making it possible to assess the degree of disparity between populations in terms of their immune responses to SARS-CoV-2, and to identify contributing factors. Scientists have identified around 900 genes that that respond differently to SARS-CoV-2 between populations. Using statistical genetic analyses, they show that these disparities are mainly due to variation in blood cellular composition: the proportion of each cell type differs from one population to another. We know that blood cell composition can be influenced by environmental factors such as exposure to cytomegalovirus (a human infection of the herpes family, which is usually harmless) and cytomegalovirus prevalence varies widely among populations: Central Africans present 99% seropositivity, in contrast to only 50% in East Asians and 32% in Europeans. The team found that an individual's environment, specifically latent cytomegalovirus infection, will thus influence the immune cell response to SARS-CoV-2. Furthermore, the scientists have identified around 1,200 human genes whose expression in response to SARS-CoV-2 is under the control of human genetic factors and the frequency of the alleles that regulate these genes can vary between the populations studied. Using population genetics approaches, they have identified recurrent selection events targeting genes involved in anti-viral functions. "We know that infectious agents have had a strong impact on human survival and exerted massive selective pressures that have shaped population genetic variation. We show that past natural selection has impacted present immune responses to SARS-CoV-2, particularly in people of East Asian ancestry, in whom coronaviruses generated strong selective pressures around 25,000 years ago," explains Maxime Rotival, a researcher in the Institut Pasteur's Human Evolutionary Genetics Unit and co-last author of the study. Between 1.5% and 2% of the genomes of Europeans and Asians is of Neanderthal origin. There is growing evidence of links between Neanderthal ancestry and present-day immunity to infection. By comparing the 1,200 genes identified with the Neanderthal genome, the scientists have discovered dozens of genes that both alter antiviral mechanisms and result from ancient introgression between Neanderthals and modern humans (Homo sapiens). "Previous studies have shown the link between some of the genes identified in our study and the severity of COVID-19. Our comprehensive population-based study highlights the direct impact of genetic variants governing immune responses to SARS-CoV-2 on the severity of COVID-19. It also establishes links between past evolutionary events, such as natural selection or Neanderthal admixture, and current population disparities in immune responses and disease risk," explains Quintana-Murci, who is also Professor at the Collège de France and co-last author of the study. "By identifying the precise cellular and molecular pathways of the genetic variants associated with COVID-19 severity, this study paves the way for precision medicine strategies that could either identify high-risk individuals or facilitate the development of new treatments," adds Darragh Duffy, Head of the Institut Pasteur's Translational Immunology Unit. More information: Yann Aquino et al, Dissecting human population variation in single-cell responses to SARS-CoV-2, Nature (2023). DOI: 10.1038/s41586-023-06422-9 Journal information: Nature www.nature.com/articles/s41586-023-06422-9 |
|
|
|
Post by Admin on Aug 28, 2023 21:18:44 GMT
Dissecting human population variation in single-cell responses to SARS-CoV-2 Abstract Humans display substantial interindividual clinical variability after SARS-CoV-2 infection1,2,3, the genetic and immunological basis of which has begun to be deciphered4. However, the extent and drivers of population differences in immune responses to SARS-CoV-2 remain unclear. Here we report single-cell RNA-sequencing data for peripheral blood mononuclear cells—from 222 healthy donors of diverse ancestries—that were stimulated with SARS-CoV-2 or influenza A virus. We show that SARS-CoV-2 induces weaker, but more heterogeneous, interferon-stimulated gene activity compared with influenza A virus, and a unique pro-inflammatory signature in myeloid cells. Transcriptional responses to viruses display marked population differences, primarily driven by changes in cell abundance including increased lymphoid differentiation associated with latent cytomegalovirus infection. Expression quantitative trait loci and mediation analyses reveal a broad effect of cell composition on population disparities in immune responses, with genetic variants exerting a strong effect on specific loci. Furthermore, we show that natural selection has increased population differences in immune responses, particularly for variants associated with SARS-CoV-2 response in East Asians, and document the cellular and molecular mechanisms by which Neanderthal introgression has altered immune functions, such as the response of myeloid cells to viruses. Finally, colocalization and transcriptome-wide association analyses reveal an overlap between the genetic basis of immune responses to SARS-CoV-2 and COVID-19 severity, providing insights into the factors contributing to current disparities in COVID-19 risk. Main A notable feature of the COVID-19 pandemic is the substantial clinical variation among individuals infected with SARS-CoV-2, ranging from asymptomatic infection to fatal disease1,2,3. Risk factors include advanced age1 as well as male sex5, comorbidities6 and host genetics4,7,8. Furthermore, variation in innate immunity9,10,11—including inborn errors or neutralizing auto-antibodies against type I interferons12,13,14—contribute to variation in clinical outcome, and epidemiological and genetic data suggest differences between populations6,7,15,16. This, together with reports of ancestry-related differences in transcriptional responses to immune challenges17,18,19, calls for investigations of the magnitude and drivers of variation in immune responses to SARS-CoV-2 across populations worldwide. Pathogen-imposed selection pressures have been paramount during human evolution20. Human adaptation to RNA viruses, through selective sweeps or archaic admixture, has been identified as a source of population genetic differentiation18,21,22 and adaptation signals have been reported at coronavirus-interacting proteins in East Asians23,24. There is also evidence for links between archaic introgression and immunity25, with Neanderthal haplotypes associated with COVID-19 severity26,27. However, the effects of natural selection and archaic admixture on immune responses to SARS-CoV-2 remain to be investigated. We addressed these questions by exposing peripheral blood mononuclear cells (PBMCs) from individuals of Central African, West European and East Asian descent to SARS-CoV-2 and, for comparison, to influenza A virus (IAV). By combining single-cell RNA-sequencing (scRNA-seq) with quantitative and population genetics approaches, we delineate environmental and genetic drivers of population differences in immune responses to SARS-CoV-2. Single-cell responses to RNA viruses We characterized transcriptional responses to SARS-CoV-2 and IAV by performing scRNA-seq analysis of PBMCs from 222 SARS-CoV-2-naive donors originating from three geographical locations (Central Africa, n = 80 male; West Europe, n = 80 male; East Asia, n = 36 female and 26 male) and with different genetic ancestries (Supplementary Fig. 1 and Supplementary Table 1). PBMCs were treated for 6 h (Supplementary Note 1, Supplementary Fig. 2 and Supplementary Table 2) with a mock-control (non-stimulated), SARS-CoV-2 (ancestral strain, BetaCoV/France/GE1973/2020) or IAV (H1N1/PR/8/1934). We captured over 1 million high-quality single-cell transcriptomes (Fig. 1a, Supplementary Fig. 3 and Supplementary Table 3a). By combining transcriptome-based clusters with cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq; Methods), we defined 22 cell types across myeloid, B, CD4+ T, CD8+ T and natural killer (NK) immune lineages (Fig. 1b, Supplementary Fig. 4 and Supplementary Table 3b–d). After virus exposure, most cell types showed moderate changes in abundance, with the strongest changes observed in the myeloid lineage after IAV treatment (Supplementary Note 2 and Supplementary Table 3e). Fig. 1: Population single-cell responses to SARS-CoV-2 and IAV. 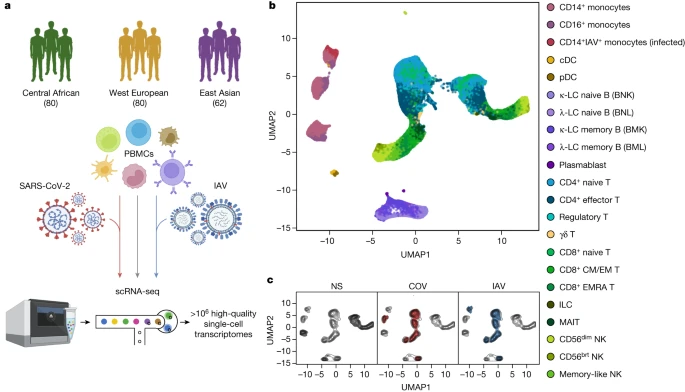 a, The study design. The diagram was created using BioRender. b,c, Uniform manifold approximation and projection (UMAP) embedding of 1,047,824 PBMCs: resting (non-stimulated; NS) or stimulated with SARS-CoV-2 (COV) or IAV for 6 h. b, The colours indicate the 22 cell types inferred. c, The distribution of cells in the NS, COV and IAV conditions on UMAP coordinates. The contour plot indicates the overall density of cells, and the coloured areas delineate regions of high cell density in each condition (NS (grey), COV (red) and IAV (blue)). After adjusting for technical factors (Methods and Supplementary Fig. 5), we found that lineage identity was the main driver of gene expression variation (around 32%), followed by virus exposure (around 27%) (Fig. 1b,c). Both viruses induced a strong transcriptional response, with 2,914 genes upregulated (false-discovery rate (FDR) < 0.01, log2[FC] > 0.5; out of 12,655 with detectable expression; Supplementary Table 3f). These responses were highly correlated across lineages and featured a strong induction of interferon-stimulated genes (ISGs) (Extended Data Fig. 1a). However, myeloid responses were markedly heterogeneous, with SARS-CoV-2 inducing a transcriptional network enriched in inflammatory-response genes (Gene Ontology (GO): 0006954; fold-enrichment (FE) = 3.4, FDR < 4.9 × 10−8; Supplementary Table 3g). For example, IL1A, IL1B and CXCL8 were highly and specifically upregulated in response to SARS-CoV-2 (log2[FC] > 2.8, FDR < 2.3 × 10−36), consistent with in vitro and in vivo studies28,29. To assess interindividual variability in the response to viruses, we summarized each individual’s response as a function of their mean ISG expression (Supplementary Table 3h). SARS-CoV-2 induced more variable ISG activity than IAV across lineages30, with myeloid cells displaying the strongest differences (Levene test, P < 6.2 × 10−6; Extended Data Fig. 1b). We determined the contributions of the various interferons (IFNs) to variation of ISG activity using single-molecule arrays (SIMOA) to quantify the levels of secreted IFNα, IFNβ and IFNγ. In the SARS-CoV-2 condition, IFNα accounted for up to 57% of ISG variability (Extended Data Fig. 2a,b), consistent with its determinant role in COVID-19 pathogenesis13. IFNA1-21 transcripts were mostly produced by infected CD14+ monocytes and plasmacytoid dendritic cells (pDCs) after IAV stimulation, whereas pDCs were the only important source of IFNA1-21 after SARS-CoV-2 stimulation (that is, producing 88% of transcripts; Extended Data Fig. 2c). IFNA1-21 expression by pDCs was weaker after stimulation with SARS-CoV-2 (log2[FC] = 6.4 versus 12.5 for IAV, Wilcoxon’s rank-sum test, P = 1.2 × 10−16). Nevertheless, patterns of interindividual variability for ISG activity were notably similar after virus treatment (r = 0.60, Pearson’s P < 1.2 × 10−22; Extended Data Fig. 2d), indicating that the IFN-driven response is largely shared between SARS-CoV-2 and IAV. |
|

