|
|
Post by Admin on Feb 6, 2017 20:23:34 GMT
 Figure 1. Principal component analysis with genome sequence data. (a) Sanganji Jomon and 1000 Genomes Project worldwide humans based on 68 542 SNPs with PB. (b) Sanganji Jomon and 1000 Genomes Project East Eurasians based on 46 158 SNPs with PB. PCA When worldwide populations are compared, PCA illustrates the genetic similarity of Sanganji Jomon and East Eurasians compared with African, European, Sahulian and Native American peoples (Figure 1a, Supplementary Figures S7-S11). However, Sanganji Jomon is located slightly closer to the center of the three major population groups in Figure 1a. This indicates the genetic uniqueness of Sanganji Jomon among East Eurasians, and/or the effect of post-mortem changes in ancient DNA, although the latter is unlikely because the result using only transversion sites showed very similar results (Supplementary Figures S8 and S10). Next, we investigated the genetic relationship between the Sanganji Jomon and East Eurasians. Comparison with 1000 Genomes Project24 East Asians (JPT (Japanese Tokyo), CHB (Han Chinese in Beijing), CHS (Southern Han Chinese), CDX (Chinese Dai in Xishuangbanna, China) and KHV (Kinh in Ho Chi Minh City, Vietnam)) based on 46 158 SNP sites show that the Sanganji Jomon is located quite apart from the other modern East Eurasians, and modern Japanese are situated between the Sanganji Jomon and continental East Eurasians (Figure 1b). Although the Sanganji Jomon data were merged for sequence data of A1 and B, the PCA plot location of the Sanganji Jomon was similar to Figure 1b when A1 and B were independently analyzed (Supplementary Figure S13). We therefore surmise that the merged data are reliable for further analyses. The uniqueness of the Sanganji Jomon was also observed when only transversion sites were used (Supplementary Figure S12), again indicating that the uniqueness was not the result of post-mortem changes. The comparison with the genome-wide SNP data of HGDP populations27 also showed the unique status of the Sanganji Jomon, who was positioned far apart from all modern East Eurasians in PC2 and PC3, although only 6864 SNPs were used. (Figure 2a,Supplementary Figure S14). The uniqueness of the Sanganji Jomon within East Eurasians is consistent with the results including Europeans and Africans. When the Ainu, the mainland Japanese and the Ryukyuan from the Japanese Archipelago1 and CHB28 were compared with Sanganji Jomon, PC1 separated the Ainu and Sanganji Jomon from the other populations (Figure 2b). The population closest to the Sanganji Jomon was the Ainu, followed by the Ryukyuan and then the mainland Japanese. It appears that PC1 corresponds to the degree of genetic contribution from the Jomon people to the other Japanese Archipelago populations, whereas PC2 separated the Ainu from Sanganji Jomon. When the genomic data of 1000 Genomes Project East Asians24 were included in the PCA analysis (Figure 2b), PC3 separated the Ainu from the Sanganji Jomon (Supplementary Figure S15).  Figure 2. Principal component analysis with genome-wide SNP data. (a) Sanganji Jomon and HGDP East Eurasians based on 6864 SNPs with PB. (b) Sanganji Jomon, individuals of three populations inhabiting the Japanese Archipelago (Ainu, mainland Japanese and Ryukyuan), and Chinese Beijing (CHB) based on 5392 SNP sites with PB. Allele sharing analysis Allele sharing analysis using 5392 SNP sites (Figure 3a) showed that the Ainu had the highest percentage of allele sharing with the Sanganji Jomon, followed by the Ryukyuan, the mainland Japanese and CHB, similar to the projection of PC1 in Figure 2b. Using the HGDP East Eurasian data set with 7081 SNP sites (Figure 3b), the mainland Japanese had the highest allele sharing with the Sanganji Jomon. Interestingly, southern East Eurasians (green bars) had slightly higher allele-sharing percentages than northern East Eurasians (blue bars), although we have to be careful with the effect of post-mortem changes.  Figure 3. Allele sharing between Sanganji Jomon and modern humans. Vertical line indicates the frequency of having the same allele with the Sanganji Jomon. (a) Comparison with Japanese Archipelago populations and Chinese Beijing (CHB) based on 5392 SNP sites with PB, (b) comparison with HGDP East Eurasians, Sahulians and Native Americans based on 7081 SNP sites with PB. Colors blue, green, brown and violet indicate northern East Eurasians, southern East Eurasians, Sahulians and Native Americans, respectively. |
|
|
|
Post by Admin on Feb 7, 2017 20:21:41 GMT
 Figure 4. Phylogenetic analysis I. TreeMix tree with gene flows. A comparison of Sanganji Jomon, 1000 Genomes Project worldwide populations, Papuan, Karitiana, Mal’ta MA1, Ust’-Ishim and Denisovan based on 43 310 all sites. Denisovan was used as the outgroup, and three gene flow events were estimated. The tree was drawn by using MEGA6.38 Red colored values (only those higher than 90% are shown) are bootstrap probabilities (%) for their adjacent internal branch. Arrows were manually added to this tree, and colors of migration weight (ratio of gene flow) follow TreeMix outputs. Values inside arrows are the ratio of gene flow. Bootstrap probabilities (%) of the gene flow from Sanganji Jomon to JPT, Karitiana to the root of European and Mal’ta MA1, and LWK to European, estimated out of 1000 bootstrap replicate TreeMix outputs, are 42%, 86% and 0.4%, respectively. TreeMix analysis We constructed maximum-likelihood population trees (ML tree) using TreeMix to investigate the phylogenetic relationship and the presence of admixture events between Sanganji Jomon and other human populations. When three migration events were assumed, the gene flow from Sanganji Jomon to JPT (modern Japanese) appeared (Figure 4) (bootstrap probability=42%). The directionality of this gene flow event was as expected, however, the inferred gene flow from Karitiana to Mal’ta MA1 (Supplementary Results S2; Supplementary Figures S16b-j and S17c-j), was in the reverse direction to what was reported by Raghavan et al.23 who used a much larger sequence data. This result might have been caused by using a relatively small SNP data set. We therefore used only modern human data, and reran TreeMix. We used a much larger 0.7 million SNP loci data. However, the resulting tree showed some anomalous gene flow directions; from Papuans to Denisovans (bootstrap probability=98%) and CEU to Papuans (bootstrap probability=86%), whereas the tree topology was consistent with that of Figure 4 (see Supplementary Figure S19). The reason for this anomaly may be that some filtering steps between our analyses and Reich et al.16 and Meyer et al.19 are different, and/or homozygous diploid genotypes in individual genome (that is, Denisovan, Papuan and so on) were used instead of their original genotype, though gene flow from Mal’ta MA1 to Karitiana correctly appeared in both all sites and transversion only when we add Mal’ta MA1, Karitiana and Ust’-Ishim to the large SNP loci data (data not shown). Figure 4.  Figure 5. Phylogenetic analysis II. Distance-based. A total of 15 519 transversion sites were used for computing distances. (a) Neighbor-joining tree. (b) Neighbor-net network. Branches for populations starting with ‘~’ were shortened for clarity. Distance matrix-based analysis We constructed neighbor-joining trees (Figure 5a and Supplementary Figure S20) from distance matrices, and the Native American diverged after the divergence of the Sanganji Jomon from the modern East Eurasians in these trees. This is consistent with Figure 4, although the bootstrap probability to support this branching pattern was only 64% in Figure 5a. We also drew a Neighbor-Net network using the same distance matrix (Figure 5b and Supplementary Figure S21). Major splits are consistent with the neighbor-joining tree (Figure 5a), and split X clustered the Sanganji Jomon and JPT. This split confirms the admixed nature of the mainland Japanese inferred from the PCA and TreeMix analyses. See Supplementary Results S3 for more detailed analysis using Neighbor-Net. |
|
|
|
Post by Admin on Feb 8, 2017 20:20:40 GMT
 Figure 6. D-statistic tests of Sanganji Jomon and worldwide humans. (a) Comparison of Sanganji Jomon, 1000 Genomes Project worldwide populations, Mal’ta MA1 and Ust’-Ishim based on 15 549 transversion sites. (b) Comparison with HGDP East Eurasians and Native Americans and SGVP Malay population based on 7439 SNPs. The genetic relationship between Sanganji Jomon and the archaic humans were then investigated. Genetic affinities between Neanderthal and non-African, and between Denisovan and Sahulian were previously reported.15, 16, 19, 22 The test of ((Sanganji Jomon, San), Altai Neanderthal) implies genetic affinity of the Jomon people with Neanderthals, but the degree was not much different from the other non-Africans (Supplementary Figure S31). Compared with the other East Eurasians, Sanganji Jomon did not show additional similarities with the Denisovans. PCA and phylogenetic analyses revealed the genetic relationships between the Sanganji Jomon and the modern human populations that we compared. We found that Sanganji Jomon was genetically quite distinct from the other modern East Eurasians. Our Jomon genotypes are still mostly based on a single read per SNP site, whereas the 1000 Genomes Project data24 had at least 4–6 × coverage. We need to be careful about the effect of genotype error rate even if we use only transversion sites (error rate=0.0129%). However, PCA plots using all sites (Figure 1 and Supplementary Figures S7 and S9) and those using transversion only (Supplementary Figures S8) are more or less the same, and TreeMix trees using all sites (Figure 4 and Supplementary Figures S16 and S18) and those using transversion only (Supplementary Figure S17) also showed similar positions for the Sanganji Jomon. Therefore, we believe that the phylogenetic relationship of the Sanganji Jomon can still be reliably estimated with our data. 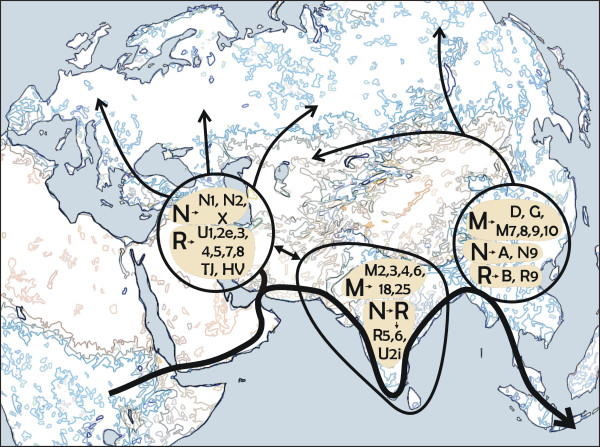 Some cranial metric and dental analyses suggested that the ancestral population of the Jomon people originated from Southeast Asia,3, 39, 40, 41, 42, 43 whereas other morphological analyses44, 45 and mtDNA sequence data9 suggested that their ancestors were of Northeast Asian origin. However, neither hypothesis was supported from our analysis using direct Jomon nuclear genome sequences. Our results suggest that the Jomon people were descendants of an ancestral East Eurasian population prior to population diversifications recognizable today. We also examined whether the uniqueness of the Sanganji Jomon in East Eurasia was the result of gene flow from non-East Eurasians, but the evidence for this was not suggested (Supplementary Figures S22, S24 and S25). This indicates that the ancestor of the Sanganji Jomon had been genetically isolated from continental populations after their divergence. However, our results do not deny a possibility of small fraction of gene flow between the Jomon people and the non-East Eurasians, which might not be detected in the current study because of the small amount of the determined Jomon genome sequences. The TreeMix tree and the D-statistic tests also confirm the lack of non-East Eurasian contaminations in the Jomon data via reagents because if there were such contamination during experiments, the effect would be artificially detected as gene flow in those analyses. Genetic affinity between modern East Eurasians and Melanesian compared with Sanganji Jomon was inferred, but these results were also ambiguous because of the data limitation. Further genomic sequencing of Jomon genomes in future studies will clarify such questions. Journal of Human Genetics (2017) 62, 213–221; doi:10.1038/jhg.2016.110; published online 1 September 2016 |
|
|
|
Post by Admin on Sept 15, 2017 19:09:45 GMT
Ishigaki Island is one of the westernmost islands in Japan. Due to its geographical location, it is considered to have played a significant role in the migration route from Southern Asia to the Japanese archipelagos. Recently, human remains were excavated from Shiraho-Saonetabaru Cave, constituting the first physical evidence of human occupation on Ishigaki Island. In order to investigate the genetic makeup of the ancient Ishigaki people and to assess their genetic relationship with other Asian populations at a molecular level, we analysed the single nucleotide polymorphisms of the coding region of mtDNA that defines the haplogroups of these individuals. Because of the poor quality of the DNA extracted from the ancient material, it was not possible to analyse all samples. Among the 10 samples considered in this study, ancient DNA data was successfully extracted from five individuals. MtDNA haplogroups show geographic specificity within Asia; the existence of haplogroup B4e and M7a in this population hints at their linkage with Southeast Asia and the Late Pleistocene Ryukyu Islands.  Figure 3.1 The geographic distribution of the islands in the Ryukyu Archipelago, and the location of Shiraho-Saonetabaru on Ishigaki Island. When ancient DNA is analysed, it is necessary to exclude false-positive results caused by contamination with contemporary DNA (Sampietro et al. 2006). In order to prevent contamination during excavation, the remains were handled using gloves and were not touched with bare hands. Bone samples were wrapped in aluminium foil and stored in a refrigerator at 4°C until DNA extraction. Standard precautions were practised to avoid contamination, such as separation of pre- and post-polymerase chain reaction (PCR) experimental setups, use of disposable laboratory gear and filter-plugged pipette tips, treatment with DNA contamination removal solution (DNA Away; Molecular Bio Products, San Diego, CA, USA), ultraviolet (UV) irradiation of equipment and benches, and negative extraction and PCR controls (Shinoda et al. 2006). DNA was extracted from the skeletal samples, according to previously published protocols (Adachi et al. 2009). The tooth and bone samples were dipped in DNA contamination removal solution for 15 min, rinsed with DNase-/RNase-free distilled water, and allowed to air-dry. The outer surface of the samples was removed using a dental drill, and the samples were again rinsed with DNase-/RNase-free distilled water and allowed to air-dry under UV irradiation. Then the tooth samples were encased in silicone rubber and the dentin around the cavitas dentis and the dental pulp was powdered and extracted through the cut plane of the root tip, as described by Gilbert et al. (2003). The bone samples were pulverised using a mill (Multi-beads shocker; Yasui Kikai, Osaka, Japan).  Figure 3.2 Results of PCR-luminex analysis. Number of each lane shows the sample number. Lane 1 and 2 are positive controls and lane 9 is negative control. A: 63 base pair amplified products indicates haplogroup M7a. B: 54 base pair products indicates haplogroup M7a1. M: size marker. Though small in number, the distribution of mtDNA haplogroups in this period provides insights into regional population history. Haplogroups B4 and R are the most prevalent in Southeast Asia, especially in the coastal region (Trejaut et al. 2005), indicating that this haplogroup may have been introduced to Japan from Southeast Asia. Interestingly, haplogroup B was also found in ancient Chinese samples (Fu et al. 2012). It seems that the ancestral population of coastal East Asia and Island Southeast Asia was enriched by the founder lineages of haplogroup B4, and the Ryukyu Islands may be one of the northernmost regions where this population arrived in the Palaeolithic period. This finding indicates that the southeast influx into the ancient Ryukyu population affected their genetic makeup and that the ancestors of the Aboriginal Taiwanese or Asian coastal region populations might be the main source of this haplogroup in the Ryukyu Islands. The geographic specificity of haplogroup M7a is the most intriguing result of this study. Its ancestral haplogroup M7, although a characteristic of East Asian populations, was not found in the northeast region of the continent (Torroni et al. 1993; Derenko et al. 2007). Haplogroup M7a is absent or scarce in the East and Southeast Asian populations outside Japan. Moreover, M7a is one of the prevailing haplogroups not only among modern Japanese, including Honshu, Okinawa islanders, and Ainu populations (Tanaka et al. 2004), but also in the Jomon population (Adachi et al. 2011). The frequency of haplogroup M7a among modern Japanese is highest in the Okinawa islanders (23.3 per cent; Umetsu et al. 2005) – gradually decreasing towards the northern part of Honshu (Shinoda 2007). This finding indicates that this haplogroup may have a southern origin. Moreover, the age of haplogroup M7a was calculated to be ca. 23,000 BP, and the age likely falls within the onset of the Last Glacial Maximum (LGM; Adachi et al. 2011).  Figure 3.3 Results of amplified product length polymorphism (APLP) analysis. Number of each lane shows the sample number. Our results confirm that the haplogroup M7a entered Japan, with the earliest settlers more than 20,000 years ago from Southeast Asia or the southern region of the Asian continent. The fact that positive proof of human occupation by haplogroup M7a on Ishigaki Island appears about 30,000–20,000 BP fits this scenario. The peopling in Japan can be seen as a complex process, as the earliest settlements and recent migrations affected the resident populations differently. Although we can draw limited conclusions from this study, our results of ancient mtDNA analysis help to shed light on late Palaeolithic human migrations to, and within, the Japanese archipelagos from Southeast Asia. Since hot and humid conditions are unfavourable for DNA preservation, there is a low possibility of finding well-preserved DNA in regions with a tropical climate, like the Ryukyu Islands. However, the present study shows that sufficient amounts of DNA were available in the human skeletal samples from the late Palaeolithic period obtained from the Ryukyu Islands because the remains were protected within caves. It seems that caves are favourable burial sites from the viewpoint of DNA preservation (Fehren-Schmitz et al. 2011). However, further studies are necessary to obtain more details on the human skeletal remains excavated from this region. Shinoda, Ken-ichi, and Noboru Adachi. “Ancient DNA Analysis of Palaeolithic Ryukyu Islanders.” New Perspectives in Southeast Asian and Pacific Prehistory, edited by Philip J. Piper et al., vol. 45, ANU Press, Acton ACT, Australia, 2017, pp. 51–60. JSTOR, www.jstor.org/stable/j.ctt1pwtd26.10. |
|
|
|
Post by Admin on Mar 26, 2018 19:00:50 GMT
 The Ainu, the indigenous people living on the northernmost island of Japan, Hokkaido (Figure 1), have long been a focus of anthropological and archeological interest because of their unique cultural, linguistic, and physical characteristics. From a cultural perspective, it is generally agreed that the ethnic identity of the Ainu was established in the 13th century AD. However, the origin of their biological identity remains unclear, which has been a major focus of anthropological discussion. Conventionally, based on the close morphological resemblance between the Jomon people and the Ainu (e.g., Brace & Nagai, 1982; Dodo & Ishida, 1990; Dodo & Kawakubo, 2002; Hanihara, 1991; Turner, 1976, 1990; Yamaguchi, 1963, 1982), it is generally agreed that the indigenous Hokkaido population maintained its genetic and phenetic characteristics throughout the Neolithic Jomon era to the Ainu era (Figure 2) (Dodo & Kawakubo, 2002). 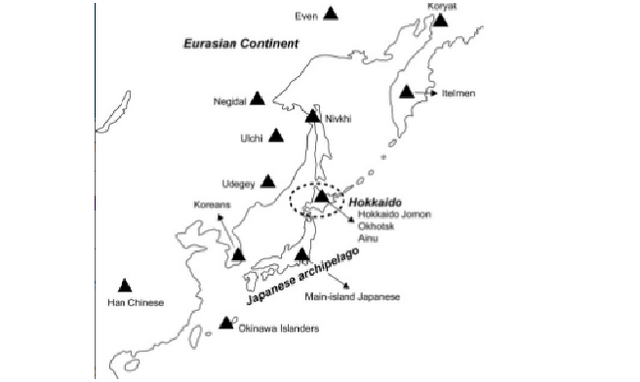 Figure 1. Map of Asia and the geographic locations of the populations analyzed or used for comparison in this study However, recent morphological and genetic studies (e.g., Hanihara, Yoshida, & Ishida, 2008; Hanihara, 2010; Ishida, Hanihara, Kondo, & Fukumine, 2009; Sato et al., 2009; Shigematsu, Ishida, Goto, & Hanihara, 2004) have indicated that the Siberian influence via the Okhotsk culture people on the Ainu significantly affected the latter's genetic structure. The Okhotsk culture people are thought to have migrated from northeastern Eurasia and been distributed in the coastal regions of northern and northeastern Hokkaido as well as southern Sakhalin during the 5th to 13th centuries AD (Amano, 2003). On Hokkaido, this culture was rapidly diminished by the invasion of Satsumon culture people at the end of the 9th century. As a result, Okhotsk culture had almost disappeared by the beginning of the 10th century. However, in the easternmost part of Hokkaido, the Okhotsk culture transformed into the Tobinitai culture under the strong influence of the Satsumon culture, and this culture continued until the beginning of the Ainu era (Segawa, 2007). Recently, we also confirmed the considerable genetic influence of the Okhotsk culture people on the formation of the modern-day Ainu. We found that mitochondrial DNA (mtDNA) haplogroups A, C, and Y, which are shared by the modern-day Siberian populations, Okhotsk culture people, and the modern-day Ainu, are not observed in the Hokkaido Jomon people (Adachi et al., 2011). However, previous genetic studies aimed at clarifying the ethnic derivation of the Ainu have two major drawbacks. The first is that almost all of these studies (Hammer et al., 2006; Harihara, Hirai, & Omoto, 1986; Harihara et al., 1988; Horai et al., 1996; Jinam et al., 2015, 2012; Tajima et al., 2004) were based on modern-day samples. Historically, after the Meiji government started sending settlers to Hokkaido as a national policy in 1869 (Fumoto, 2004), an enormous number of mainland Japanese migrated to Hokkaido, and their apparent genetic influence on the modern-day Ainu was confirmed by a genome-wide single-nucleotide polymorphism (SNP) analysis by Jinam et al. (2012). In this context, the extent of the earlier genetic contribution of the mainland Japanese to the original formation of the Ainu cannot be evaluated. 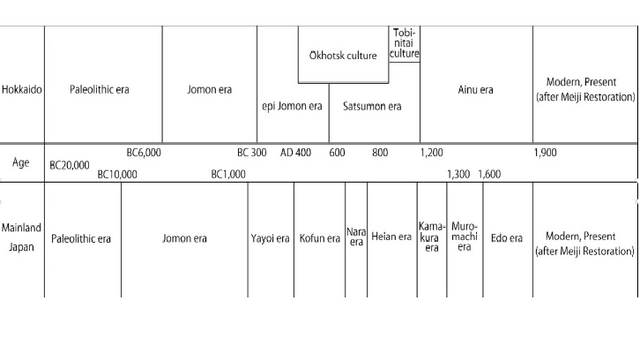 Figure 2. Chronological table of Hokkaido and mainland Japan The second problem is that the study area focused on in previous genetic studies is limited or unclear. The samples used in most studies were derived from only 51 individuals who lived in Biratori Town, central Hokkaido. Actually, Ainu people live throughout Hokkaido, so it is unclear whether these 51 individuals can represent all of the Ainu. Besides these studies, Horai et al. (1991) analyzed six early-modern (200–300 years BP) Ainu in Hokkaido. However, unfortunately, they did not describe the region of origin of these samples. This sampling bias precludes genetic evaluation of possible regional differences of the Ainu as suggested by previous morphological studies (e.g., Dodo, Kawakubo, Sawada, & Ishida, 2012; Hanihara et al., 2008; Hanihara, 2010; Ito, 1967; Kondo, 1995; Ossenberg, Dodo, Maeda, & Kawakubo, 2006; Shigematsu et al., 2004; Yamaguchi, 1981). Therefore, to clarify the process of formation of the Ainu and their regional characteristics, individuals who were not influenced by the large-scale immigration of mainland Japanese must be examined. In addition, regional bias of the samples must be corrected. In the current study, we thus present the mtDNA data of Ainu skeletons excavated from all over Hokkaido. All of these skeletons are dated to the Edo era (1603 to 1868 AD), a period for which no distinct proof of an intensive genetic influence of mainland Japanese on the Ainu has been reported. A detailed analysis of the coding and control regions of mtDNA was used to address the questions regarding the matrilineal genetic characteristics of the Ainu, as well as those about the relationship between the Ainu and the ancient and modern-day populations in eastern Eurasia. Moreover, regional differences of the Ainu were discussed. |
|

