|
|
Post by Admin on Jul 5, 2018 18:47:34 GMT
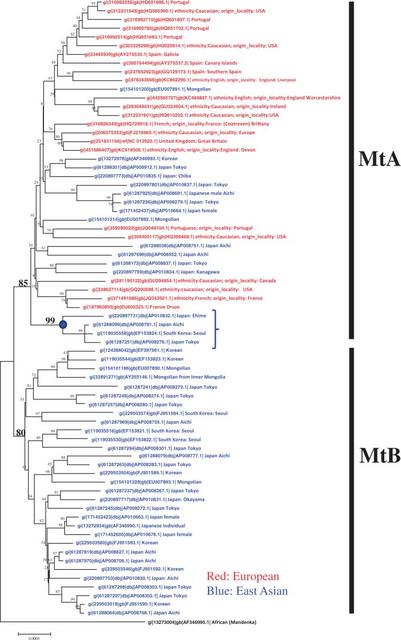 Fig. 2.— Phylogenetic tree of the complete mitochondrial genomes for the four ethnic groups. It is divided into MtA and MtB. MtA contains East Asian and European individuals together while MtB includes East Asian individuals only. For the male lineages, we used the Y-STR markers for 453 individuals covering Japanese, Korean, Mongolian (Khalkh), American, and European people. We determined the evolutionary distances (RST) among them, and constructed a phylogenetic tree using the NJ method (Saitou and Nei 1987), as shown in figure 1. The Y-STR tree revealed that the male ancestral lineage contained two clades (Yap-A and Yap-B). While Yap-A clade includes the East Asian individuals only, Yap-B clade contains the East Asian and European individuals together. Surprisingly, the European males never formed an independent clade. Instead, they formed separate clades within Yap-B. We then constructed a phylogenetic tree (Mt tree) for the 72 complete mtDNAs including the four ethnic groups, as shown in figure 2. We used Kimura’s two parameter method (Kimura 1980) for computing evolutionary distances among them and the NJ method for the tree construction. The Mt tree revealed two female descendant clades (Mt A and Mt B). Mt B consisted of the East Asian females only, while Mt A contained the East Asian and European females together. As in the case of males, the European females did not form an independent clade, but comprised several groups within the Mt A tree. As the Mt A cluster includes roughly as many European individuals as East Asian individuals, it is not clear which of them is ancestral to the other. The node marked with the blue circle in figure 2 suggests that the East Asians are ancestral to the Europeans. The bootstrap value of the node is 99%. Therefore, both male and female lineages suggest that Europeans diverged from within East Asian ancestors or that they interbred with East Asian individuals up to a certain divergence time.  Our next question was thus to estimate the divergence time of the European clade within the East Asian lineage, for males and females separately. To address that question, we computed the evolutionary distance (RST) between every pair of the male individuals to construct the Y-STR tree in figure 1. The RST value between the bottom and root of A and B clades in the tree was 16.91, while that between the bottom and root of the European male clade in the B clade was 12.31. Based on a divergence of East Asians from Africans of 55,000 years ago (Nei and Roychoudhury 1974, 1993), and assuming that RST is proportional to time, we can estimate the evolutionary rate of Y-STR by using the Y-STR tree. In the tree the RST value between the deepest root and the bottom is 16.91, and that between the common ancestor of Europeans males and the bottom is 12.31. The rate is thus estimated as 16.91/55,000 = 3.07 × 10−4 per repeat per year, which leads us to the conclusion that the divergence time of the European males is ∼40,100 years ago. In the case of the Mt tree, we first transformed it into tree in which the lengths of a pair of branch lengths from the common node were equal to one another, as in the UPGMA tree (Sokal 1958), because we now dealt with the evolutionary time rather than distance (Kumada 1993). In the transformed tree, the topology was kept unchanged. We call the transformed tree the evolutionary time (ET) tree (fig. 3), because the tree reflects the evolutionary time rather than the distance, we call the tree showing evolutionary distance the evolutionary distance (ED) tree. |
|
|
|
Post by Admin on Jul 7, 2018 18:47:42 GMT
 Fig. 3.— A phylogenetic tree of the complete mitochondrion genome (16,750 bp) including D-loop regions of 72 humans; 32 Japanese, 11 Korean, 5 Mongolian, 23 European, and 1 African samples. In the present ET tree, the distance between the bottom and root in any route was 0.00132 per nucleotide site, while that between the bottom and the root (marked with the closed circle) of the European male clade was 0.000996. Therefore, assuming again that the two major clades in the Mt tree diverged from the African ∼55,000 years ago, we estimated the evolutionary rate of mtDNA as 2.4 × 10−8 per site per year. Our estimate was comparable with that of Ingman et al. (2000). It is noted that our estimate for the human mitochondrial genome is more than ten times faster than that of the human nuclear neutral sequences (Fukami Kobayashi 2005). Then, by applying the evolutionary rate of mtDNA to the distance between the bottom and root in the European clade, we estimated the divergence time of the European females, as ∼41,500 years ago. Thus, both the Y-STR and Mt trees are mutually consistent with respect to the tree shape and divergence time of European individuals relative to East Asians, despite being based on independent data and distance measures. Our results suggest that the European people settled down in their territories ∼41,000 year ago, and have developed their own cultures and languages since then. On the other hand, the East Asians were classified into two clusters; one is Type 1 East Asians denoted as Yap-A and Mt B clusters in the male and female trees, respectively, and the other is Type 2 East Asians denoted as Yap-B and Mt A clusters in the male and female trees, respectively. While Type 1 includes East Asian individuals only, Type 2 contains East Asians and European individuals together. The general view of the East Asian and European divergences is summarized in figure 4.  Fig. 4.— Phylogenetic tree of the major human populations Africans, Type 1 East Asians, Europeans, and Type 2 East Asians. Both the Y-STR and mtDNA trees consistently show that Europeans diverged from East Asian ancestors ∼41,000 years ago. Population genetic theory indicates that 41,000 years, or about 2,000 generations, are long enough to accumulate SNPs in the same loci in each lineage (Kimura 1983; Nei 1987) to account for the present genetic and phenotypic differences between the East Asians and Europeans, but too short to acquire independent loci between them. Recently, Liu (2012) reported on five genes responsible for the facial morphology of European people. The East Asian people must have the counterparts that differ at the SNP level from those in the European people. As our phylogenetic trees demonstrate, the European alleles at the five loci have diverged from the ancestral East Asian alleles. Our result contrasts with the traditional view that Europeans and East Asians simultaneously diverged from African ancestors 55,000 years ago. It is noteworthy, however, that Shinoda (2007) investigated into the haplogroups of mtDNA, and revealed a number of evolutionary haplotype lineages. The lineages include L3 (African), N (East Asian), W (European), and L3 to N to R (East Asian) and then HV (European) among others. Though they did not explain their results, their haplotype lineages can now be understood by our finding that the Europeans diverged from the East Asians. Therefore, the discrepancy between the traditional view and ours lies mainly in that the traditional view was based on autosomal genes that evolved much slower than Y-STR or mtDNA, and could not distinguish the evolutionary lineages at the individual level. Note that, as estimated earlier, the evolutionary rate of mtDNA is 2.4 × 10−8 per site per year, while that of nuclear neutral sequences is 2.0 × 10−9 per site per year (Fukami Kobayashi 2005). The discrepancy also is due to the fact that while we dealt with many male and female individuals in our study, the other studies did not. By typing the 14 binary markers of the Y-STR sequences according to the classification agreed at the Y Chromosome Consortium (2002) and elsewhere (Karafet et al. 2008), we found that all individuals in the Yap-B1 in figure 1 belonged to either haplogroup C or D, while the majority in the Yap- B2 belonged to haplogroup O. Since Yap-B1 includes mainly Japanese and Korean males, in which 91% individuals share haplotype O2b, the Korean and Japanese males are definitely closest to one another within East Asian humans. We also found a high frequency of the O2b haplotype in Manchu (Northern China) and Korean-Chinese samples (Katoh et al. 2005b; Kim et al. 2011). The origin of the Ainu people is still an unresolved issue (Tajima et al. 2004). On the basis of our results, we propose a possible scenario for the origin of the Ainu people, who now live in the north-most island of the Japanese archipelago, Hokkaido. The Ainu people have European phenotypic characters, but they are genetically closer to East Asians than to Europeans (Watanabe 1975). These contradictory features of the Ainu people are puzzling. As shown in figure 4, Europeans may have diverged from East Asians ∼41,000 years ago, it is possible that hybrid individuals were born before the divergence, and some of them looked more like the Europeans while possessing a generally East Asian genotype. We suggest that the ancestor of the Ainu people was such a group of the hybrid individuals. We note that the present Ainu people share the mtDNA haplotype not with the Japanese but with the European living in Siberia, Russia (Adachi et al. 2009). Thus, we furthermore suggest that the ancestor of the Ainu originated in northern Eurasia and took a route through Siberia and north China before settling in northern regions of Japan and nearby places. There is a report that other people than the Ainu also lived in the northern regions but disappeared (Adachi et al. 2009). As the people in Okinawa islands are closest to the Ainu people in the East Asians (Jinam et al. 2012), they might also be descendants from of mixing of East Asian and European lineages. Genome Biol Evol. 2014 Mar; 6(3): 466–473. |
|
|
|
Post by Admin on Aug 20, 2018 18:25:09 GMT
Genome-wide single-nucleotide polymorphism (SNP) data analyses1, 2 support the dual-structure model3, 4 for the formation of modern Japanese populations, in which indigenous Jomon people admixed with later migrants who brought rice agriculture. The Jomon culture geographically ranged from Hokkaido to the Okinawa islands, and the Jomon people inhabited the Japanese Archipelago from ~16000 to 2500 years before present (YBP).5, 6 The origins and phylogenetic relationships of the Jomon people, however, are still elusive. Ancient DNA sequences of the Jomon people provide direct evidence of their genetic characteristics. Mitochondrial DNA (mtDNA) sequences of the Jomon people and their haplotypes have been determined for many individuals.7, 8, 9, 10, 11, 12 Haplogroup N9b, whose frequency is generally low in modern East Eurasians,13, 14 was found to be quite frequent in the mtDNA of the Jomons.9, 10, 11 This suggests a long-term isolation of the Jomons from continental populations. However, inferring human population history only from mtDNA data are insufficient because of their limited genetic information. Thanks to new technologies, it is now possible to analyze nuclear genome sequences of ancient human remains15, 16, 17, 18, 19, 20, 21, 22, 23 and those of modern human individuals.24 We therefore determined the nuclear genome sequences of two Jomon individuals and compared them with available data so as to infer the origin of modern Japanese.  Figure 1 Principal component analysis with genome sequence data. (a) Sanganji Jomon and 1000 Genomes Project worldwide humans based on 68 542 SNPs with PB. (b) Sanganji Jomon and 1000 Genomes Project East Eurasians based on 46 158 SNPs with PB. Genetic relationship between Sanganji Jomon and other modern and archaic humans We investigated the genetic relationships between Sanganji Jomon and modern humans from different parts of the world, including Africans, West Eurasians, East Eurasians, Native Americans and Sahulians37 (descendants of people who migrated to Sahul land); see the map shown in Supplementary Figure S1.  Figure 2 Principal component analysis with genome-wide SNP data. (a) Sanganji Jomon and HGDP East Eurasians based on 6864 SNPs with PB. (b) Sanganji Jomon, individuals of three populations inhabiting the Japanese Archipelago (Ainu, mainland Japanese and Ryukyuan), and Chinese Beijing (CHB) based on 5392 SNP sites with PB. PCA When worldwide populations are compared, PCA illustrates the genetic similarity of Sanganji Jomon and East Eurasians compared with African, European, Sahulian and Native American peoples (Figure 1a, Supplementary Figures S7-S11). However, Sanganji Jomon is located slightly closer to the center of the three major population groups in Figure 1a. This indicates the genetic uniqueness of Sanganji Jomon among East Eurasians, and/or the effect of post-mortem changes in ancient DNA, although the latter is unlikely because the result using only transversion sites showed very similar results (Supplementary Figures S8 and S10). Next, we investigated the genetic relationship between the Sanganji Jomon and East Eurasians. Comparison with 1000 Genomes Project24 East Asians (JPT (Japanese Tokyo), CHB (Han Chinese in Beijing), CHS (Southern Han Chinese), CDX (Chinese Dai in Xishuangbanna, China) and KHV (Kinh in Ho Chi Minh City, Vietnam)) based on 46 158 SNP sites show that the Sanganji Jomon is located quite apart from the other modern East Eurasians, and modern Japanese are situated between the Sanganji Jomon and continental East Eurasians (Figure 1b). Although the Sanganji Jomon data were merged for sequence data of A1 and B, the PCA plot location of the Sanganji Jomon was similar to Figure 1b when A1 and B were independently analyzed (Supplementary Figure S13). We therefore surmise that the merged data are reliable for further analyses. The uniqueness of the Sanganji Jomon was also observed when only transversion sites were used (Supplementary Figure S12), again indicating that the uniqueness was not the result of post-mortem changes.The comparison with the genome-wide SNP data of HGDP populations27 also showed the unique status of the Sanganji Jomon, who was positioned far apart from all modern East Eurasians in PC2 and PC3, although only 6864 SNPs were used. (Figure 2a,Supplementary Figure S14). The uniqueness of the Sanganji Jomon within East Eurasians is consistent with the results including Europeans and Africans. When the Ainu, the mainland Japanese and the Ryukyuan from the Japanese Archipelago1 and CHB28 were compared with Sanganji Jomon, PC1 separated the Ainu and Sanganji Jomon from the other populations (Figure 2b). The population closest to the Sanganji Jomon was the Ainu, followed by the Ryukyuan and then the mainland Japanese. It appears that PC1 corresponds to the degree of genetic contribution from the Jomon people to the other Japanese Archipelago populations, whereas PC2 separated the Ainu from Sanganji Jomon. When the genomic data of 1000 Genomes Project East Asians24 were included in the PCA analysis (Figure 2b), PC3 separated the Ainu from the Sanganji Jomon (Supplementary Figure S15). |
|
|
|
Post by Admin on Aug 22, 2018 18:16:15 GMT
 Figure 3 Allele sharing between Sanganji Jomon and modern humans. Vertical line indicates the frequency of having the same allele with the Sanganji Jomon. (a) Comparison with Japanese Archipelago populations and Chinese Beijing (CHB) based on 5392 SNP sites with PB, (b) comparison with HGDP East Eurasians, Sahulians and Native Americans based on 7081 SNP sites with PB. Colors blue, green, brown and violet indicate northern East Eurasians, southern East Eurasians, Sahulians and Native Americans, respectively. Allele sharing analysis Allele sharing analysis using 5392 SNP sites (Figure 3a) showed that the Ainu had the highest percentage of allele sharing with the Sanganji Jomon, followed by the Ryukyuan, the mainland Japanese and CHB, similar to the projection of PC1 in Figure 2b. Using the HGDP East Eurasian data set with 7081 SNP sites (Figure 3b), the mainland Japanese had the highest allele sharing with the Sanganji Jomon. Interestingly, southern East Eurasians (green bars) had slightly higher allele-sharing percentages than northern East Eurasians (blue bars), although we have to be careful with the effect of post-mortem changes. TreeMix analysis We constructed maximum-likelihood population trees (ML tree) using TreeMix to investigate the phylogenetic relationship and the presence of admixture events between Sanganji Jomon and other human populations. When three migration events were assumed, the gene flow from Sanganji Jomon to JPT (modern Japanese) appeared (Figure 4) (bootstrap probability=42%). The directionality of this gene flow event was as expected, however, the inferred gene flow from Karitiana to Mal’ta MA1 (Supplementary Results S2; Supplementary Figures S16b-j and S17c-j), was in the reverse direction to what was reported by Raghavan et al.23 who used a much larger sequence data. This result might have been caused by using a relatively small SNP data set. We therefore used only modern human data, and reran TreeMix. We used a much larger 0.7 million SNP loci data. However, the resulting tree showed some anomalous gene flow directions; from Papuans to Denisovans (bootstrap probability=98%) and CEU to Papuans (bootstrap probability=86%), whereas the tree topology was consistent with that of Figure 4 (see Supplementary Figure S19). The reason for this anomaly may be that some filtering steps between our analyses and Reich et al.16 and Meyer et al.19 are different, and/or homozygous diploid genotypes in individual genome (that is, Denisovan, Papuan and so on) were used instead of their original genotype, though gene flow from Mal’ta MA1 to Karitiana correctly appeared in both all sites and transversion only when we add Mal’ta MA1, Karitiana and Ust’-Ishim to the large SNP loci data (data not shown).  Figure 4 Phylogenetic analysis I. TreeMix tree with gene flows. A comparison of Sanganji Jomon, 1000 Genomes Project worldwide populations, Papuan, Karitiana, Mal’ta MA1, Ust’-Ishim and Denisovan based on 43 310 all sites. Denisovan was used as the outgroup, and three gene flow events were estimated. The tree was drawn by using MEGA6.38 Red colored values (only those higher than 90% are shown) are bootstrap probabilities (%) for their adjacent internal branch. Arrows were manually added to this tree, and colors of migration weight (ratio of gene flow) follow TreeMix outputs. Values inside arrows are the ratio of gene flow. Bootstrap probabilities (%) of the gene flow from Sanganji Jomon to JPT, Karitiana to the root of European and Mal’ta MA1, and LWK to European, estimated out of 1000 bootstrap replicate TreeMix outputs, are 42%, 86% and 0.4%, respectively. |
|
|
|
Post by Admin on Aug 23, 2018 18:45:40 GMT
Distance matrix-based analysis We constructed neighbor-joining trees (Figure 5a and Supplementary Figure S20) from distance matrices, and the Native American diverged after the divergence of the Sanganji Jomon from the modern East Eurasians in these trees. This is consistent with Figure 4, although the bootstrap probability to support this branching pattern was only 64% in Figure 5a. We also drew a Neighbor-Net network using the same distance matrix (Figure 5b and Supplementary Figure S21). Major splits are consistent with the neighbor-joining tree (Figure 5a), and split X clustered the Sanganji Jomon and JPT. This split confirms the admixed nature of the mainland Japanese inferred from the PCA and TreeMix analyses. See Supplementary Results S3 for more detailed analysis using Neighbor-Net.  Figure 5 Phylogenetic analysis II. Distance-based. A total of 15 519 transversion sites were used for computing distances. (a) Neighbor-joining tree. (b) Neighbor-net network. Branches for populations starting with ‘∼’ were shortened for clarity. D-statistic analysis In agreement with the PCA and TreeMix results, the D-statistic test also suggested genetic affinity of the Sanganji Jomon with the East Eurasians, especially with the mainland Japanese, compared with non-East Eurasians (Figure 6a and b). This reflects the genetic continuity between the Jomon and the mainland Japanese, and support the hypothesis of an early divergence of the Jomon with the ancestor of modern East Eurasians (Supplementary Figure S23). See also Supplementary Results S4 for additional analyses using D-statistics.  Figure 6 The genetic relationship between Sanganji Jomon and the archaic humans were then investigated. Genetic affinities between Neanderthal and non-African, and between Denisovan and Sahulian were previously reported.15, 16, 19, 22The test of ((Sanganji Jomon, San), Altai Neanderthal) implies genetic affinity of the Jomon people with Neanderthals, but the degree was not much different from the other non-Africans (Supplementary Figure S31). Compared with the other East Eurasians, Sanganji Jomon did not show additional similarities with the Denisovans. |
|

