Post by Admin on Feb 21, 2019 18:40:02 GMT
Allele sharing analysis
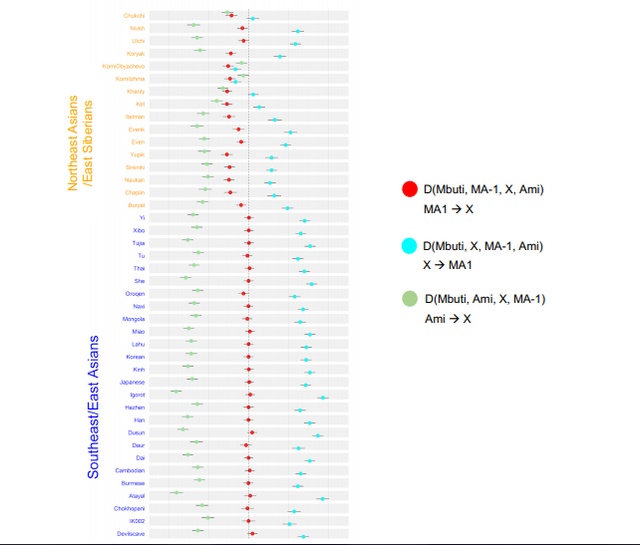
Allele sharing analysis using 5392 SNP sites (Figure 3a) showed that the Ainu had the highest percentage of allele sharing with the Sanganji Jomon, followed by the Ryukyuan, the mainland Japanese and CHB, similar to the projection of PC1 in Figure 2b. Using the HGDP East Eurasian data set with 7081 SNP sites (Figure 3b), the mainland Japanese had the highest allele sharing with the Sanganji Jomon. Interestingly, southern East Eurasians (green bars) had slightly higher allele-sharing percentages than northern East Eurasians (blue bars), although we have to be careful with the effect of post-mortem changes.

Figure 3
Allele sharing between Sanganji Jomon and modern humans. Vertical line indicates the frequency of having the same allele with the Sanganji Jomon. (a) Comparison with Japanese Archipelago populations and Chinese Beijing (CHB) based on 5392 SNP sites with PB, (b) comparison with HGDP East Eurasians, Sahulians and Native Americans based on 7081 SNP sites with PB. Colors blue, green, brown and violet indicate northern East Eurasians, southern East Eurasians, Sahulians and Native Americans, respectively.
TreeMix analysis
We constructed maximum-likelihood population trees (ML tree) using TreeMix to investigate the phylogenetic relationship and the presence of admixture events between Sanganji Jomon and other human populations. When three migration events were assumed, the gene flow from Sanganji Jomon to JPT (modern Japanese) appeared (Figure 4) (bootstrap probability=42%). The directionality of this gene flow event was as expected, however, the inferred gene flow from Karitiana to Mal’ta MA1 (Supplementary Results S2; Supplementary Figures S16b-j and S17c-j), was in the reverse direction to what was reported by Raghavan et al.23 who used a much larger sequence data. This result might have been caused by using a relatively small SNP data set. We therefore used only modern human data, and reran TreeMix. We used a much larger 0.7 million SNP loci data. However, the resulting tree showed some anomalous gene flow directions; from Papuans to Denisovans (bootstrap probability=98%) and CEU to Papuans (bootstrap probability=86%), whereas the tree topology was consistent with that of Figure 4 (see Supplementary Figure S19). The reason for this anomaly may be that some filtering steps between our analyses and Reich et al.16 and Meyer et al.19 are different, and/or homozygous diploid genotypes in individual genome (that is, Denisovan, Papuan and so on) were used instead of their original genotype, though gene flow from Mal’ta MA1 to Karitiana correctly appeared in both all sites and transversion only when we add Mal’ta MA1, Karitiana and Ust’-Ishim to the large SNP loci data (data not shown).

Figure 4
Phylogenetic analysis I. TreeMix tree with gene flows. A comparison of Sanganji Jomon, 1000 Genomes Project worldwide populations, Papuan, Karitiana, Mal’ta MA1, Ust’-Ishim and Denisovan based on 43 310 all sites. Denisovan was used as the outgroup, and three gene flow events were estimated. The tree was drawn by using MEGA6.38 Red colored values (only those higher than 90% are shown) are bootstrap probabilities (%) for their adjacent internal branch. Arrows were manually added to this tree, and colors of migration weight (ratio of gene flow) follow TreeMix outputs. Values inside arrows are the ratio of gene flow. Bootstrap probabilities (%) of the gene flow from Sanganji Jomon to JPT, Karitiana to the root of European and Mal’ta MA1, and LWK to European, estimated out of 1000 bootstrap replicate TreeMix outputs, are 42%, 86% and 0.4%, respectively.
Allele sharing analysis using 5392 SNP sites (Figure 3a) showed that the Ainu had the highest percentage of allele sharing with the Sanganji Jomon, followed by the Ryukyuan, the mainland Japanese and CHB, similar to the projection of PC1 in Figure 2b. Using the HGDP East Eurasian data set with 7081 SNP sites (Figure 3b), the mainland Japanese had the highest allele sharing with the Sanganji Jomon. Interestingly, southern East Eurasians (green bars) had slightly higher allele-sharing percentages than northern East Eurasians (blue bars), although we have to be careful with the effect of post-mortem changes.
Figure 3
Allele sharing between Sanganji Jomon and modern humans. Vertical line indicates the frequency of having the same allele with the Sanganji Jomon. (a) Comparison with Japanese Archipelago populations and Chinese Beijing (CHB) based on 5392 SNP sites with PB, (b) comparison with HGDP East Eurasians, Sahulians and Native Americans based on 7081 SNP sites with PB. Colors blue, green, brown and violet indicate northern East Eurasians, southern East Eurasians, Sahulians and Native Americans, respectively.
TreeMix analysis
We constructed maximum-likelihood population trees (ML tree) using TreeMix to investigate the phylogenetic relationship and the presence of admixture events between Sanganji Jomon and other human populations. When three migration events were assumed, the gene flow from Sanganji Jomon to JPT (modern Japanese) appeared (Figure 4) (bootstrap probability=42%). The directionality of this gene flow event was as expected, however, the inferred gene flow from Karitiana to Mal’ta MA1 (Supplementary Results S2; Supplementary Figures S16b-j and S17c-j), was in the reverse direction to what was reported by Raghavan et al.23 who used a much larger sequence data. This result might have been caused by using a relatively small SNP data set. We therefore used only modern human data, and reran TreeMix. We used a much larger 0.7 million SNP loci data. However, the resulting tree showed some anomalous gene flow directions; from Papuans to Denisovans (bootstrap probability=98%) and CEU to Papuans (bootstrap probability=86%), whereas the tree topology was consistent with that of Figure 4 (see Supplementary Figure S19). The reason for this anomaly may be that some filtering steps between our analyses and Reich et al.16 and Meyer et al.19 are different, and/or homozygous diploid genotypes in individual genome (that is, Denisovan, Papuan and so on) were used instead of their original genotype, though gene flow from Mal’ta MA1 to Karitiana correctly appeared in both all sites and transversion only when we add Mal’ta MA1, Karitiana and Ust’-Ishim to the large SNP loci data (data not shown).
Figure 4
Phylogenetic analysis I. TreeMix tree with gene flows. A comparison of Sanganji Jomon, 1000 Genomes Project worldwide populations, Papuan, Karitiana, Mal’ta MA1, Ust’-Ishim and Denisovan based on 43 310 all sites. Denisovan was used as the outgroup, and three gene flow events were estimated. The tree was drawn by using MEGA6.38 Red colored values (only those higher than 90% are shown) are bootstrap probabilities (%) for their adjacent internal branch. Arrows were manually added to this tree, and colors of migration weight (ratio of gene flow) follow TreeMix outputs. Values inside arrows are the ratio of gene flow. Bootstrap probabilities (%) of the gene flow from Sanganji Jomon to JPT, Karitiana to the root of European and Mal’ta MA1, and LWK to European, estimated out of 1000 bootstrap replicate TreeMix outputs, are 42%, 86% and 0.4%, respectively.