|
|
Post by Admin on Dec 25, 2020 19:54:03 GMT
Denisovan gene flow into the ancestors of Melanesians Although the Denisova individual derives from a population that was not directly involved in the gene flow from Neanderthals to Eurasians, it is possible that Denisovans admixed with the ancestors of present-day people in some parts of the Old World. To investigate this, we analysed the relationship of the Denisova genome to the genomes of 938 present-day humans from 53 populations that have been genotyped at 642,690 single nucleotide polymorphisms (SNPs)35. We scored each of these present-day humans based on their relative proximity to Neanderthals and the Denisova individual at positions where we have high-quality data for both the Neanderthal and Denisova genomes (Supplementary Information section 8). Using the means of the 53 populations, the first two principal components separate the populations into three groups (Fig. 2): first, the 7 sub-Saharan African populations; second, a group of 44 non-African populations as well as one north African group; and third, Papuan and Bougainville populations from Melanesia. When individuals from selected populations are analysed separately, the Papuan and Bougainville islanders remain distinct from almost all individuals outside Africa (Supplementary Fig. 8.1b). Thus, with respect to their relationship to Neanderthals and Denisovans, the Melanesian populations stand out relative to other non-African populations. Figure 2: Relationship of present-day populations to the Denisova individual and Neanderthals based on 255,077 SNPs. 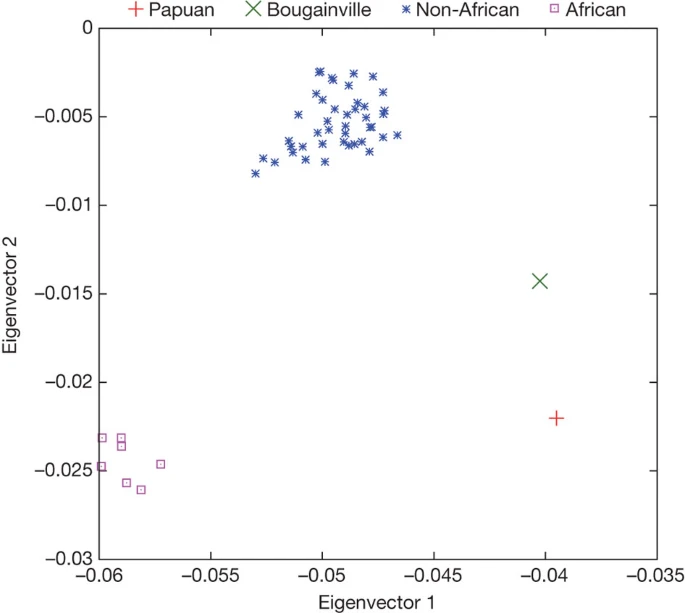 Principal component analysis of the means of 53 present-day human populations projected onto the top two principal components defined by Denisova, Neanderthal and chimpanzee. The seven ‘African’ populations are San, Mbuti, Biaka, Bantu Kenya, Bantu South Africa, Yoruba and Mandenka; the ‘Non-African’ populations are 44 diverse groups from outside Africa except for Papuan and Bougainville islanders. To explore this further, we analysed the relationship of the Denisova genome to the genomes of five present-day humans that we previously sequenced to about fivefold coverage8 (a Yoruba and a San genome from Africa, a French genome from Europe, a Han genome from China and a Papuan genome from Melanesia), as well as seven present-day humans that we sequenced to 1–2-fold coverage for this study (a Mbuti genome from Africa, a Sardinian genome from Europe, a Mongolian genome from Central Asia, a Cambodian genome from South-East Asia, an additional Papuan genome from Melanesia, a Bougainville islander genome from Melanesia, and a Karitiana genome from South America) (Supplementary Information section 9). We used the D statistic8 to test if various pairs of present-day humans share equal numbers of derived alleles with the Denisova individual. To do this, we restricted comparisons to pairs of present-day humans sequenced at the same time to minimize the chance that differences in sample processing could affect the results. We find that the fivefold coverage Papuan individual shares 4.0 ± 0.7% more alleles with the Denisova individual than does the French individual, and we observed a similar skew in all 10 comparisons of Melanesian and other non-African populations (Table 1). When we stratified the data by base substitution class and chromosome, the D statistics are qualitatively unchanged (Supplementary Information section 10). Similarly, the D statistics are consistent for all depths of read coverage, indicating that mapping errors, for example due to segmental duplications, are not likely to explain these results. Finally, differences in sequencing error rate across samples cannot explain the observed D statistics (Supplementary Information section 10). Under the assumption that gene flow explains these observations, we determined the direction of this gene flow by asking whether Melanesians and other Eurasians share derived alleles with Africans equally often. If the gene flow was entirely into the ancestors of the Denisovan individual, we would not expect this to affect the relationship of Africans to Melanesians and other Eurasians and thus we would expect them to share derived alleles equally often with Africans. However, we find that derived alleles in Africans match Melanesians 3.4 ± 0.4% less often than other non-Africans (Z = 10.8). Because this skew is seen without using Denisovan data it cannot be explained by gene flow into Denisovans or, for example, by contamination of the Denisova sample by present-day Melanesian DNA. Thus, at least some of the putative gene flow must have been into Melanesians (Supplementary Information section 8). When we compare the skew in the fraction of derived alleles shared with the two archaic hominins to what would be expected for individuals of 100% Neanderthal or Denisova ancestry, respectively (Supplementary Information section 8 and ref. 8), we estimate that 2.5 ± 0.6% of the genomes of non-African populations derive from Neanderthals, in agreement with our previous estimate of 1–4%8. In addition, we estimate that 4.8 ± 0.5% of the genomes of Melanesians derive from Denisovans. Altogether, as much as 7.4 ± 0.8% of the genomes of Melanesians may thus derive from recent admixture with archaic hominins. |
|
|
|
Post by Admin on Dec 26, 2020 20:31:35 GMT
A model of population history To understand the implications of the relationships observed among the Denisova individual, the Neanderthals and present-day humans, we fit the D statistics described in the previous sections to a parameterized model of population history. The D statistics for the Denisova individual differ in two important ways from those for the Neanderthal. First, the Denisova individual shares fewer derived alleles with either the French or Han Chinese populations than do the Neanderthals. Second, the Denisova individual shares more derived alleles with the Papuans than do the Neanderthals. We are able to fit the data with a model that assumes the Denisovans are a sister group of Neanderthals with a population divergence time of one-half to two-thirds of the time to the common ancestor of Neanderthals and humans. After the divergence of the Denisovans from Neanderthals, there was gene flow from Neanderthals into the ancestors of all present-day non-Africans. Later there was admixture between the Denisovans and the ancestors of Melanesians that did not affect other non-African populations. This model is illustrated in Fig. 3 and is described in detail in Supplementary Information section 11. Figure 3: A model of population history compatible with the data. 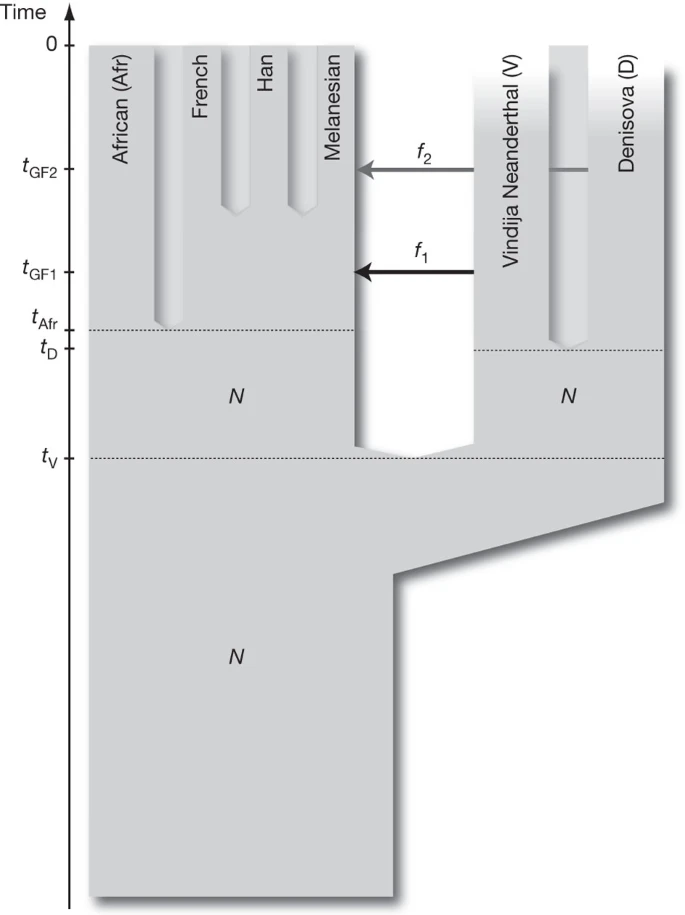 Other, more complex models could also explain the data. For example, a model that invokes only gene flow from Denisovans to Melanesian ancestors outside Africa and assumes four subpopulations in Africa that existed between the times of the origin of Denisovan and Neanderthal ancestors and the ancestors of present-day Eurasians could also fit the data (Supplementary Fig. 11.4). However, because barriers to gene flow between such subpopulations would have to persist for hundreds of thousands of years to create the observed patterns, such a model is less plausible on biological grounds than a model that invokes two instances of gene flow outside Africa. |
|
|
|
Post by Admin on Dec 27, 2020 21:17:11 GMT
A tooth from Denisova Cave In 2000, a hominin tooth was discovered in layer 11.1 of the south gallery of Denisova Cave (Fig. 4a, b). The tooth is from a young adult and therefore from another individual than the phalanx which stems from a juvenile (Supplementary Information section 12). To elucidate the relationship of the tooth to the individual from which the phalanx is derived, we extracted DNA from 50 mg of dentin from the root of the tooth and prepared a sequencing library (Supplementary Information section 13). About 0.17% of random DNA sequences determined from this library aligned to the human genome, whereas the rest is likely to represent microbial contamination common in ancient bones. We therefore used a novel DNA capture approach36 to isolate mtDNA sequences from the sequencing library. A total of 15,094 sequences were identified which allowed the complete mtDNA genome to be assembled at an average coverage of 58-fold. This sequence differs at two positions from the mtDNA of the phalanx whereas it differs at about 380 positions from both Neanderthal and present-day humans. The time since the most recent common ancestor of the two mtDNAs from Denisova Cave is estimated to be 7,500 years, with a 95% upper bound of 16,000 years (Supplementary Information section 13). We conclude that the tooth and the phalanx derive from two different individuals that are probably from the same hominin population. Figure 4: Morphology of the Denisova molar. 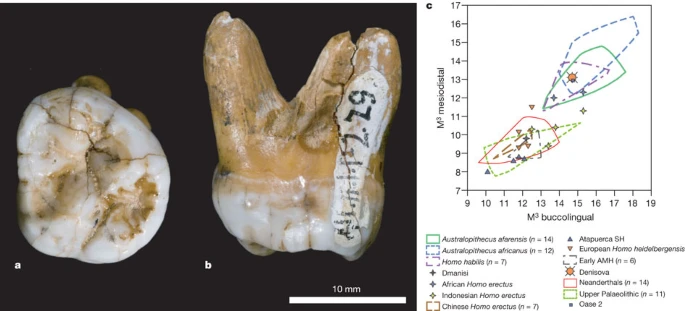 a, b, Occlusal (a) and mesial (b) views. c, Comparison of the Denisova molar to diverse third molars, in a biplot of the mesiodistal and buccolingual lengths (in mm). AMH, anatomically modern humans; SH, Sima de los Huesos. Supplementary Fig. 12.1 presents a similar comparison to second molars. Morphology of the Denisova molar The tooth is an almost complete left, probably third, but possibly second, upper molar (Fig. 4b). The crown is trapezoidal and tapers strongly distally, with bulging lingual and buccal walls giving the tooth an inflated appearance (Supplementary Information section 12). The roots are short but robust and strongly flaring. Overall, the tooth is very large (mesiodistal diameter, 13.1 mm; buccolingual, 14.7 mm). As a third molar, it is outside the range of normal size variation of all fossil taxa of the genus Homo, with the exception of H. habilis and H. rudolfensis, and comparable to Australopithecines (Fig. 4c). Compared to second molars, it is larger than Neanderthals or early modern humans, but similar to H. erectus and H. habilis (Supplementary Fig. 12.1). Besides size, it is also distinguished from most Neanderthal third molars by the absence of hypocone reduction, and from both second and third Neanderthal molars by the presence of a large talon basin and the strong flare of the crown. Furthermore, it lacks the lingual hypocone projection seen in all Neanderthal first and many second molars, and has strongly diverging roots, unlike the closely spaced and frequently fused roots of Neanderthals. It is of particular interest to compare the Denisova molar to Middle Pleistocene hominins from China, where H. erectus and other archaic forms, sometimes interpreted as H. heidelbergensis, may have survived until recently. Unfortunately, very few of these fossils preserve third upper molars. Of the few examples that are available, most differ from the Denisova molar by their strongly reduced size. Second molars are more frequent than third molars, and most have a trapezoidal shape like Denisova, but they do not have the lingually skewed position of the hypocone and metacone and the strong basal flare of the crown. The Denisova molar supports the DNA evidence that the Denisovan population is distinct from late Neanderthals as well as from modern humans. In fact, the primitive traits of the Denisova tooth suggest that Denisovans may have been separated from the Neanderthal lineage before Neanderthal dental features are documented in Western Eurasia (>300,000 years BP) (Supplementary Information section 12), although we cannot exclude the possibility that the Denisovan dental morphology results from a reversion. Stratigraphy and dating The small size of both the phalanx and the tooth precludes direct radiocarbon dating. We instead dated seven bone fragments found close to the hominin remains in layer 11 in the east and south galleries. To ensure that they were associated with human occupation of the cave we chose bones that have evidence of human modification, including a rib with regular incisions and a bone projectile point blank generally associated with Upper Palaeolithic cultural assemblages. In the south gallery, where modified bones were not available, we used herbivore bones (Supplementary Information section 12). Four of the seven dates are infinite dates older than 50,000 years BP (uncalibrated), whereas three are finite dates between 16,000 and 30,000 years BP (Supplementary Table 12.1). The rib with incisions and the projectile point blank are about 30,000 and 23,000 years BP, respectively. Together with three previous dates23 this shows that layer 11 contains cultural remains from at least two different time periods, one period older than 50,000 years BP and one more recent period. However, the stratigraphy is complicated by the discovery of a wedge-shaped area close to the area where the phalanx was found that is likely to be disturbed (Supplementary Information section 12). Hominin remains large enough to allow direct radiocarbon dates may eventually be discovered in the cave, but a reasonable hypothesis is that the phalanx and molar belong to the older occupation. |
|
|
|
Post by Admin on Dec 28, 2020 5:42:45 GMT
Discussion
The molecular preservation of the Denisova phalanx is exceptional in that the fraction of endogenous relative to microbial DNA is about 70%. By contrast, in all Neanderthal remains studied so far the relative abundance of endogenous DNA is below 5%, and typically below 1%. Furthermore, the average length of hominin DNA fragments in the Denisova phalanx is 58 base pairs (bp) (SL3003) and 74 bp (SL3004) in spite of the enzymatic treatment that removes uracil residues and decreases the average fragment size, whereas in most well-preserved Neanderthal samples it is 50 bp or smaller without this treatment. Thus, although many Neanderthals are preserved under conditions apparently similar to those in Denisova Cave, the Denisova phalanx is one of few bones found in temperate conditions that are as well preserved as many permafrost remains37,38. It is not clear why this is. It is not due to some condition that affects all hominin remains in Denisova Cave because the fraction of endogenous DNA in the tooth is 0.17%; that is, typical of other Late Pleistocene hominin remains. It is possible that a rapid desiccation of the tissue after death, which would limit degradation of the DNA by endogenous enzymes as well as microbial growth, has allowed this exceptional preservation.
The Denisova individual and the population to which it belonged carry some exceptionally archaic molecular (mtDNA) as well as morphological (dental) features. Nevertheless, the picture that emerges from analysis of the nuclear genome is one where the Denisova population is a sister group to Neanderthals. Three possibilities could account for how such archaic features have come to be present in Denisovans. One possibility is that these features were retained in Denisovans but became lost in modern humans and Neanderthals. A second, not mutually exclusive, possibility is that they entered the Denisova population through gene flow from some even more diverged hominin. Although such gene flow cannot be detected with the current mtDNA and nuclear DNA data, further sequencing of other hominin remains may in the future allow testing for it. A third possibility that could account for the apparently archaic dental morphology, but not the mtDNA, is a reversal to ancestral traits.
After they diverged from one another, Denisovans and Neanderthals had largely separate population histories as shown by a number of observations. First, patterns of allele sharing indicate that Denisovan ancestors did not contribute genes at a detectable level to present-day people all over Eurasia whereas Neanderthals did8. Thus, Neanderthals at some point interacted with ancestors of present-day Eurasians independently of Denisovans. Second, the genetic diversity of Neanderthals across their geographical range in the last thirty or forty thousand years of their history was extremely low, indicating that they experienced one or more strong genetic bottlenecks independently of the Denisovans. Third, our results indicate that Denisovans but not Neanderthals contributed genes to ancestors of present-day Melanesians. Fourth, the dental morphology shows no evidence of any derived features seen in Neanderthals. In fact, dental remains from the Sima de los Huesos of Atapuerca, for which ages between 350,000 and 600,000 years have been proposed39,40, already carry Neanderthal-like morphological features that are not seen in the Denisova molar.
An interesting question is how widespread Denisovans were. A possibility is that they lived in large parts of East Asia at the time when Neanderthals were present in Europe and western Asia. One observation compatible with this possibility is that Denisovan relatives seem to have contributed genes to present-day Melanesians but not to present-day populations which currently live much closer to the Altai region such as Han Chinese or Mongolians (Table 1). Thus, they have at least at some point been present in an area where they interacted with the ancestors of Melanesians and this was presumably not in southern Siberia. Further studies of both molecular and morphological features of hominin remains across Asia should clarify how widespread Denisovans were and how they were related to archaic hominins other than Neanderthals.
The Denisova individual belongs to a hominin group that shares a common ancestor with Neanderthals but has a distinct population history. We define this group based on genomic evidence and call it Denisovans, but refrain from any formal Linnaean taxonomic designations that would indicate species or subspecies status for either Neanderthals or Denisovans. In our view, these results show that on the Eurasian mainland there existed at least two forms of archaic hominins in the Upper Pleistocene: a western Eurasian form with morphological features that are commonly used to define them as Neanderthals, and an eastern form to which the Denisova individuals belong. In the future, when more complete genomes from these and other archaic hominins will be sequenced from remains that allow more morphological features to assessed, their relationships will become even better understood. This will be an important endeavour as the emerging picture of Upper Pleistocene hominin evolution is one in which gene flow among different hominin groups was common.
|
|
|
|
Post by Admin on Jan 1, 2021 21:11:01 GMT
Mapping gene flow between ancient hominins through demography-aware inference of the ancestral recombination graph Melissa J. Hubisz ,Amy L. Williams,Adam Siepel Published: August 6, 2020 doi.org/10.1371/journal.pgen.1008895Abstract The sequencing of Neanderthal and Denisovan genomes has yielded many new insights about interbreeding events between extinct hominins and the ancestors of modern humans. While much attention has been paid to the relatively recent gene flow from Neanderthals and Denisovans into modern humans, other instances of introgression leave more subtle genomic evidence and have received less attention. Here, we present a major extension of the ARGweaver algorithm, called ARGweaver-D, which can infer local genetic relationships under a user-defined demographic model that includes population splits and migration events. This Bayesian algorithm probabilistically samples ancestral recombination graphs (ARGs) that specify not only tree topologies and branch lengths along the genome, but also indicate migrant lineages. The sampled ARGs can therefore be parsed to produce probabilities of introgression along the genome. We show that this method is well powered to detect the archaic migration into modern humans, even with only a few samples. We then show that the method can also detect introgressed regions stemming from older migration events, or from unsampled populations. We apply it to human, Neanderthal, and Denisovan genomes, looking for signatures of older proposed migration events, including ancient humans into Neanderthal, and unknown archaic hominins into Denisovans. We identify 3% of the Neanderthal genome that is putatively introgressed from ancient humans, and estimate that the gene flow occurred between 200-300kya. We find no convincing evidence that negative selection acted against these regions. Finally, we predict that 1% of the Denisovan genome was introgressed from an unsequenced, but highly diverged, archaic hominin ancestor. About 15% of these “super-archaic” regions—comprising at least about 4Mb—were, in turn, introgressed into modern humans and continue to exist in the genomes of people alive today. Author summary We present ARGweaver-D, an extension of the ARGweaver algorithm which can be applied under a user-defined demographic model including population splits and migration events. Given genome sequence data from a collection of individuals across multiple closely related populations or subspecies, ARGweaver-D can infer trees describing the genetic relationships among these individuals at every location along the genome, conditional on the demographic model. Like ARGweaver, ARGweaver-D is a Bayesian method, sampling trees from the posterior distribution in order to account for uncertainty. Using simulations, we show that ARGweaver-D can successfully identify regions introgressed from Neanderthals and Denisovans into modern humans. It is also well-powered to detect introgressed regions stemming from older gene-flow events. We apply ARGweaver-D to the genomes of two Neanderthals, a Denisovan, and two African humans. We identify 3% of the Neanderthal genome which is likely derived from gene flow from ancient humans. We also identify about 1% of the Denisovan genome that may be traced to an unsequenced archaic hominin; 15% of these regions were subsequently passed to modern humans. We find no convincing evidence that selection acted against any of these introgressed regions. Citation: Hubisz MJ, Williams AL, Siepel A (2020) Mapping gene flow between ancient hominins through demography-aware inference of the ancestral recombination graph. PLoS Genet 16(8): e1008895. doi:10.1371/journal.pgen.1008895 |
|

